
Eur Heart J 平滑肌α-肌动蛋白错义突变体通过调控细胞内胆固醇合成促进动脉粥样硬化
2023-12-28 论道心血管 论道心血管
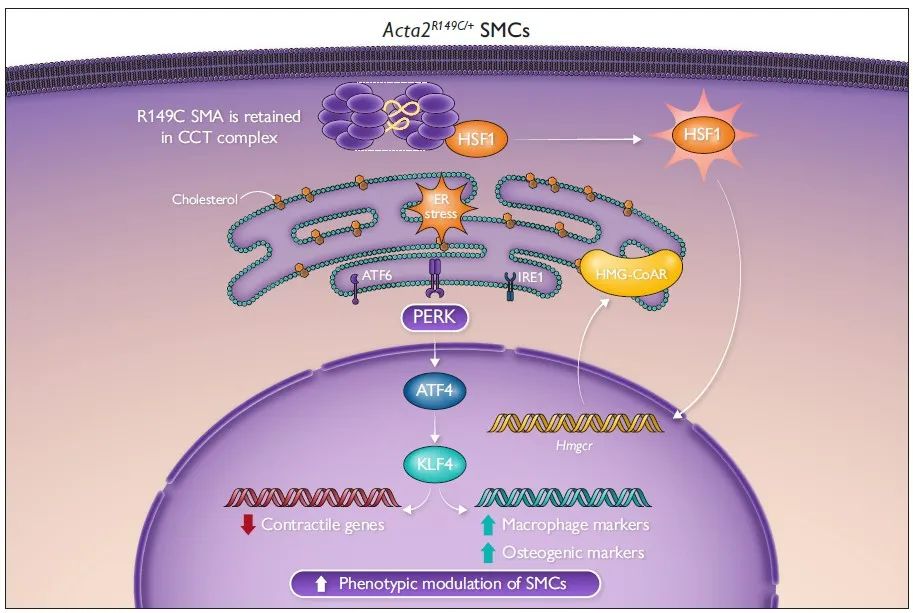
该研究发现在CCT复合体中滞留的R149C SMA可以增加内源性胆固醇的生物合成,从而诱导ER应激,激活PERK-ATF4-KLF4信号通路,进而调控SMC表型转化,促进动脉粥样硬化斑块形成。
在动脉粥样硬化斑块的形成过程中,平滑肌细胞(SMC)发生了复杂的表型转化,平滑肌细胞分化标志物(如Acta2, Cnn1)表达下调,而干细胞标志物(Ly6a)、成纤维细胞标志物(Fn1, Ecrg4)、巨噬细胞标志物(Lgals3)、软骨细胞样细胞标志物(Spp1)等则表达上调。KLF4是调控平滑肌表型转化的重要转录因子。主动脉SMCs在游离胆固醇或氧化低密度脂蛋白(oxLDL)刺激下发生表型转化,这是由于胆固醇进入内质网(ER),触发ER应激和未折叠蛋白反应(UPR)。在三种被UPR激活的内质网跨膜受体(PERK、ATF6、IRE1)中,双链RNA激活蛋白激酶样ER激酶(PERK)介导下游KLF4激活以及SMC表型转化。与高脂血症野生型(WT)小鼠相比,平滑肌特异性PERK敲除的高脂血症小鼠的动脉粥样硬化斑块形成减少了80%,这说明外源胆固醇诱导的PERK信号通路在SMC表型调控和斑块形成中发挥重要作用。
ACTA2编码平滑肌特异性α-肌动蛋白(SMA),其一种常见的突变形式p.Arg149Cys (R149C)在没有高胆固醇血症或其他危险因素的情况下与胸主动脉疾病和冠心病早发密切相关。所有肌动蛋白都需要与胞质中含有伴侣蛋白的T复合多肽(CCT)复合体相互作用才能获得其天然构象,而R149C SMA则因错误折叠而滞留在CCT复合体中。R149C SMA是否通过ER应激影响SMC表型转化和动脉粥样硬化斑块形成尚不清楚。
2023年6月28日,美国德克萨斯大学Dianna M Milewicz教授团队在European Heart Journal发表题为“Smooth muscle α-actin missense variant promotes atherosclerosis through modulation of intracellular cholesterol in smooth muscle cells”的研究成果,发现在CCT复合体中滞留的R149C SMA可以增加内源性胆固醇的生物合成,从而诱导ER应激,激活PERK-ATF4-KLF4信号通路,进而调控SMC表型转化,促进动脉粥样硬化斑块形成。

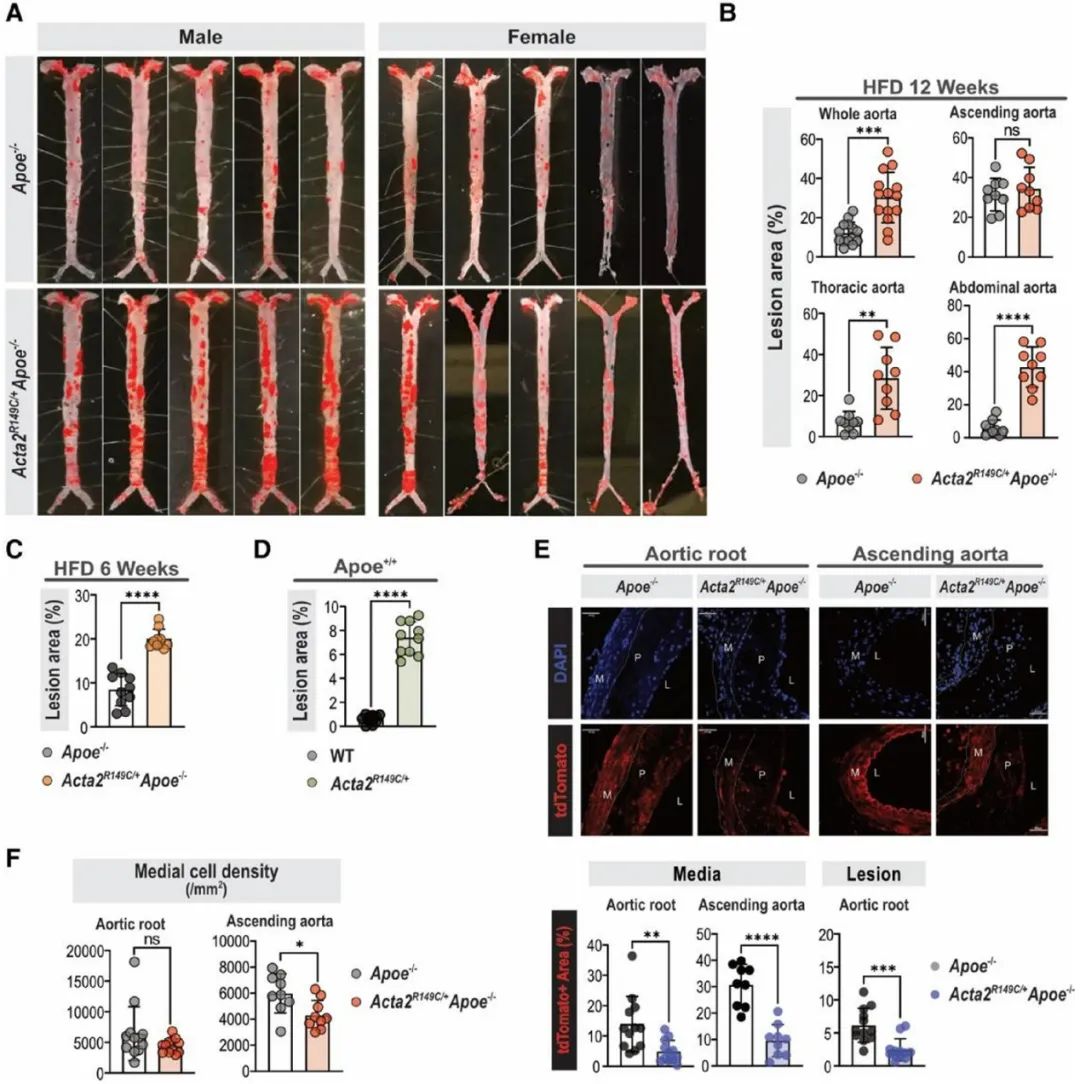
首先,研究人员构建了Acta2杂合突变(Acta2R149C/+)小鼠,并与Apoe-/-小鼠杂交获得Acta2R149C/+Apoe-/-小鼠,以Apoe-/-小鼠作为对照组。两组小鼠给予高脂饮食喂养12周构建动脉粥样硬化模型。主动脉油红O染色发现,与Apoe-/-小鼠相比,雄性和雌性Acta2R149C/+Apoe-/-小鼠的动脉粥样硬化斑块面积均增加了约2.5倍。然而,两组小鼠的血脂水平无显著差异。在没有Apoe-/-背景的情况下,高脂饮食喂养12周的Acta2R149C/+小鼠的主动脉根部和升主动脉上形成了小的动脉粥样硬化斑块,而WT小鼠的主动脉中则没有斑块形成。接下来,研究人员将Acta2R149C/+Apoe-/-及Apoe-/-小鼠与Myh11-CreERT2-tdTomatoflox/flox小鼠杂交获得平滑肌谱系示踪小鼠,发现与Apoe-/-小鼠相比,高脂饮食喂养12周的Acta2R149C/+Apoe-/-小鼠的主动脉根部斑块和中膜层tdTomato+细胞较少,而巨噬细胞较多。研究人员将上述两组小鼠主动脉弓组织进行单细胞转录组测序分析,表达tdTomato的细胞被视为SMC来源的细胞簇。全基因组关联研究已经确定了超过160个与人类冠心病风险增加相关的基因组位点,与Apoe-/-小鼠相比, Acta2R149C/+Apoe-/-小鼠主动脉的SMC在其中38个位点的表达存在显著差异,包括Klf4的表达增加。
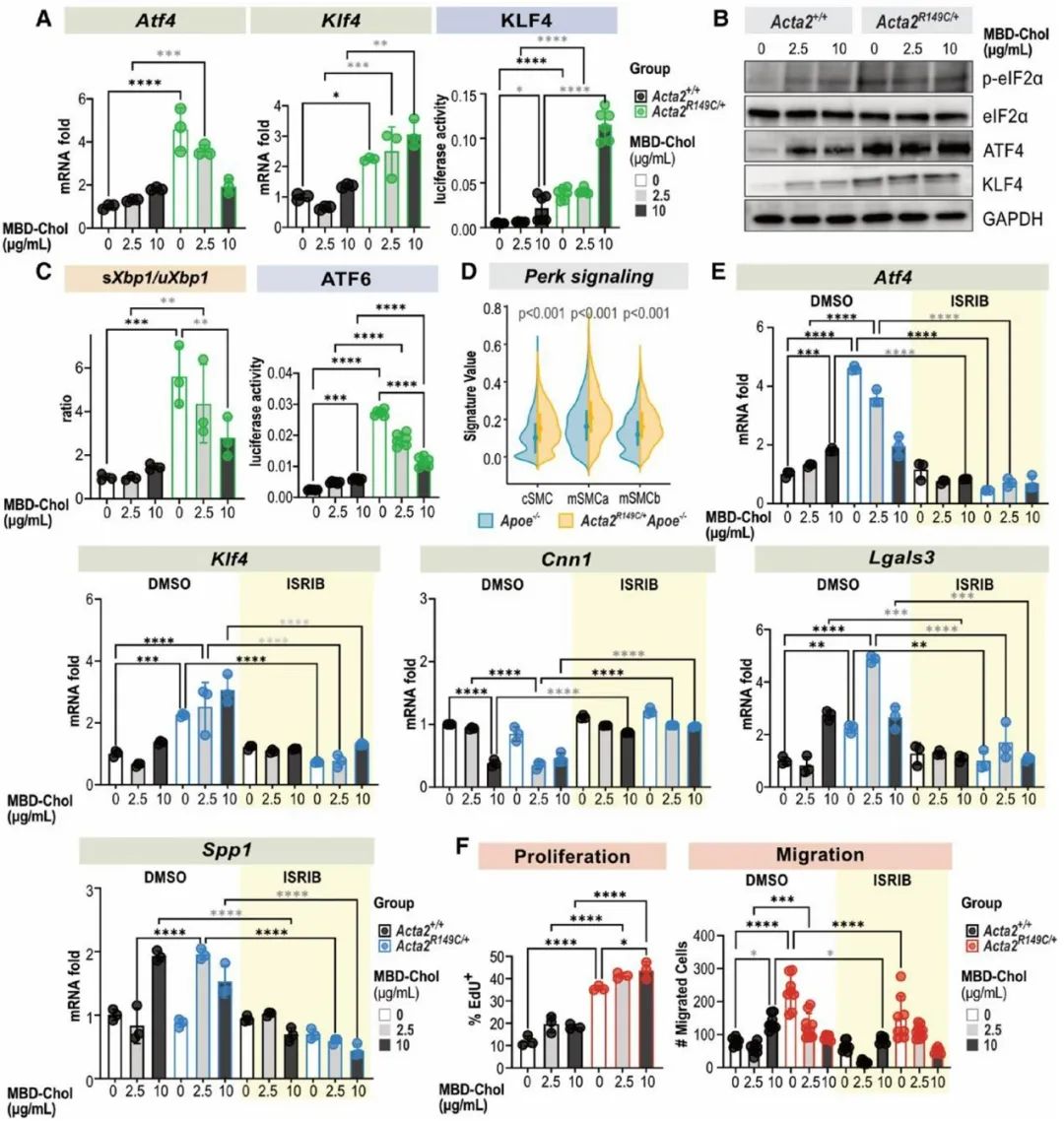
为探究SMA的R149C突变调控SMC表型转化的具体机制,研究人员提取Acta2R149C/+及WT小鼠的主动脉原代SMCs进行体外培养,并给予与甲基-β-环糊精复合的游离胆固醇(MBD-Chol)刺激72h。WT SMCs给予10μg/mL MBD-Chol刺激后三种UPR通路均被激活,而Acta2R149C/+ SMCs 在基线状态下三种UPR通路就被激活。单细胞转录组测序分析结果显示三种UPR通路中,PERK信号通路激活最为明显。同时,研究人员发现WT SMCs在给予10μg/mL MBD-Chol刺激后,才出现平滑肌收缩标志物表达减少及动脉粥样硬化相关调节基因表达增加的变化,而Acta2R149C/+ SMCs基线状态或2.5μg/mL MBD-Chol刺激下就出现上述表型转化。并且,Acta2R149C/+ SMCs增殖和迁移能力较WT SMCs显著升高。而这些变化均可被PERK抑制剂ISRIB所阻断。这些结果说明,PERK信号通路在SMA的R149C突变对SMC的表型转化调控中发挥关键作用。
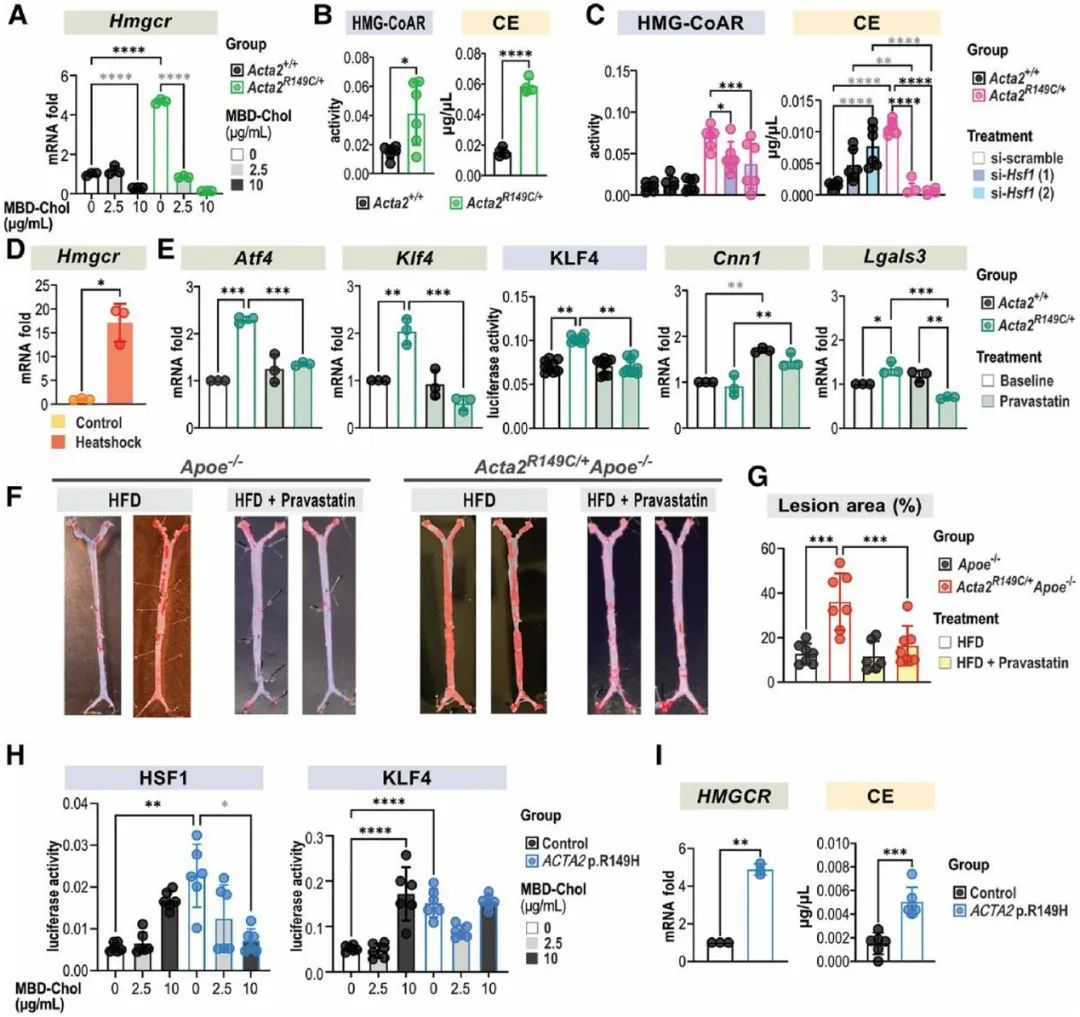
热休克因子1(HSF1)在细胞质内与CCT复合体结合,蛋白毒性应激通过磷酸化作用激活HSF1,使其富集到分子伴侣启动子的热休克反应元件上促进分子伴侣的转录。研究人员发现与WT SMCs相比,Acta2R149C/+ SMCs的HSF1、p-HSF1水平及HSF1下游靶基因表达均升高,表明突变的SMA在CCT复合体中的滞留会引发胞质应激。ER应激与胞质应激之间的相互作用已被阐明,因此,研究人员在Acta2R149C/+ SMCs中转染siHsf1,发现HSF1的敲低可以抑制PERK-ATF4-KLF4信号通路的激活,并逆转SMCs的表型转化。为了进一步证实HSF1激活引起UPR及SMC表型转化不依赖于外源性胆固醇的刺激,研究人员将SMCs在44℃热休克45分钟,并在37℃恢复12小时。热休克后,SMCs的Atf4、Klf4表达升高,同时收缩表型标志物(Cnn1)表达降低,而去分化标志物(Lgals3、Spp1)表达升高。已有文献报道HSF1激活增加胆固醇的生物合成,而内源性胆固醇和外源性胆固醇刺激同样可以引起UPR。HMG-CoAR是胆固醇生物合成的限速酶,研究人员发现相较于WT SMCs,Acta2R149C/+ SMCs的Hmgcr mRNA水平、HMG-CoAR活性及胆固醇酯水平均升高,而敲低HSF1则降低Acta2R149C/+ SMCs中HMG-CoAR活性及胆固醇酯水平。而热休克同样可以升高SMCs的Hmgcr mRNA水平。HMG-CoAR抑制剂普伐他汀可以降低Acta2R149C/+ SMCs的Atf4、Klf4表达及KLF4活性并抑制其表型转化。为了进一步证实HMG-CoAR活性增加是Acta2R149C/+Apoe-/-小鼠斑块负荷增加的原因,研究人员通过饮用水给予Acta2R149C/+Apoe-/-及Apoe-/-小鼠普伐他汀(50 mg/kg/d)治疗,该药物有效地降低了两种基因型小鼠的血脂水平,并且消除了Acta2R149C/+Apoe-/-小鼠相较于Apoe-/-小鼠的斑块负荷增加。并且,研究人员从ACTA2 p.R149H变异患者及健康对照者的主动脉提取SMCs,发现ACTA2 p.R149H SMCs相较于对照,HSF1及HMG-CoAR表达水平升高,胆固醇酯水平升高。这些结果说明,R149C SMA通过激活HSF1增加HMG-CoAR的表达及活性,从而增加细胞内胆固醇生物合成,进而引起ER应激及SMC表型转化。
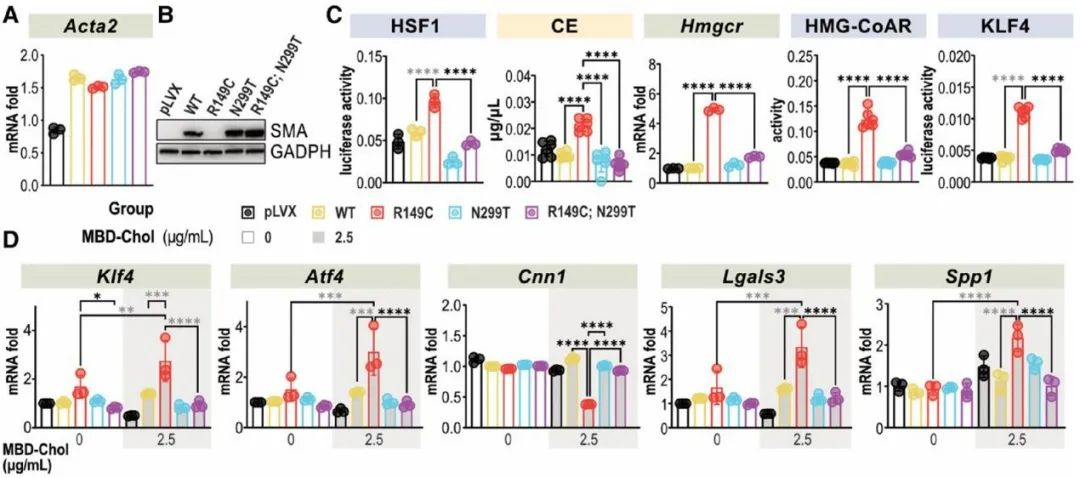
最后,研究人员探究了CCT复合体中R149C SMA的错误折叠是否与HSF1的激活及下游信号传导有关。此前研究表明,滞留在CCT复合体中R149C SMA引入一个新的突变位点(N299T)后可从复合体中释放。因此,研究人员构建了携带4种形式SMA (WT、R149C、N299T和含N299T的R149C)的慢病毒载体,转染Acta2-/- SMCs后在其中稳定表达。Acta2-/- SMCs中R149C SMA的表达可诱导HSF1激活,增加HMG-CoAR的表达、活性和胆固醇酯水平,进而激活PERK-ATF4-KLF4信号通路并引起SMCs表型转化,而R149C; N299T SMA的表达则无此作用,这说明R149C SMA在CCT复合体中的错误折叠引起了HSF1的激活。
综上所述,平滑肌细胞中R149C SMA的错误折叠可诱导HSF1介导的细胞质应激,从而增加HMG-CoAR水平和细胞内胆固醇的生物合成,细胞内胆固醇水平的升高激活了内质网应激和PERK-ATF4-KLF4信号通路,进而促进了平滑肌表型转化和动脉粥样硬化斑块形成。该研究为无高胆固醇血症的SMA突变体携带人群易患早发性冠心病这一临床现象提供了可能的机制,表明了平滑肌的遗传变异在平滑肌表型转化及动脉粥样硬化发生发展中发挥的重要作用。
原文链接:
https://academic.oup.com/eurheartj/advance-article/doi/10.1093/eurheartj/ehad373/7209229
作者:论道心血管
版权声明:
本网站所有注明“来源:梅斯医学”或“来源:MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明“来源:梅斯医学”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言







#动脉粥样硬化# #平滑肌α-肌动蛋白# #内源性胆固醇#
26