
病例分享 | 反复腹痛、水肿,疾病沉疴20余年,罪魁祸首竟是罕见HAE
2024-07-22 消化界 消化界
47 岁女性产后反复腹痛呕吐、水肿,经检查确诊遗传性血管性水肿,接受拉那利尤单抗治疗效果良好,强调早期诊断及治疗的重要性。
在时间的长河中,二十年不过是一瞬,但对于这位患者来说,却是一段漫长而煎熬的岁月。腹痛与水肿,这两个在产后便不请自来的不速之客,一次又一次侵袭着这位患者的生活。患者反复出现急性腹痛呕吐,手肿、脚背肿、整个脸也经常水肿,严重影响了她的正常生活与社交。
多次辗转各科室,这位47岁的患者终于在消化科拿到了解开自己“病痛之谜”的钥匙——遗传性血管性水肿(Hereditary angioedema, HAE)。接下来,不妨让我们跟随她就诊的足迹,看看这位患者这次究竟是如何在医生的帮助下追根溯源,找到导致自己反复腹痛、水肿的罪魁祸首的。
病例介绍
患者女性,47岁,主诉“间断上腹胀痛20余年,再发2天“。
1 病史回顾
现病史:
约23年前产后开始无明确诱因反复出现上腹部胀痛不适,进食后症状加重,偶伴恶心呕吐,无明显发作规律,应用抑酸药物可逐渐缓解。病情反复发作,曾完善胃镜检查未见异常。2天前口服钙片后再发上腹胀痛不适。患者平时常出现过敏症状,皮疹多位于肘部、膝盖、双手。
追问病史:
23年前产后开始无明确诱因反复出现双手、双足背浮肿,2-3天可自行缓解。约8年前开始无诱因反复出现面部浮肿,浮肿从额头开始向下蔓延,一般持续5-7天可自行消肿,平均每年发病3-5次,抗敏药物等均无效。
既往史:
反复过敏史,曾检测过敏原对多种食物过敏。否认其他慢性疾病病史。
家族史:
否认家族性遗传性疾病病史。
2 入院检查
急诊科体格检查:
剑突下轻压痛,余未见异常。
急诊科影像学检查:
全腹增强CT显示十二指肠管壁水肿增厚(图1)。
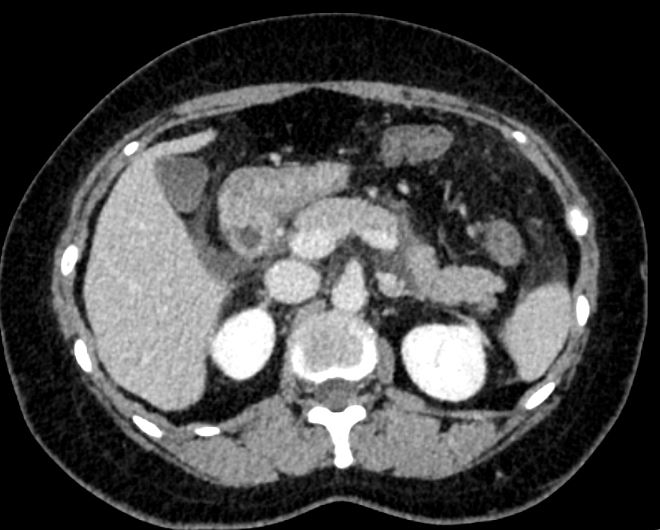
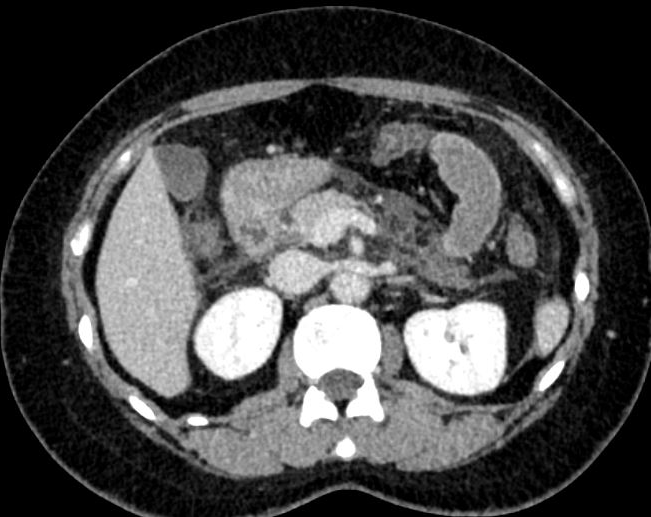
图1 全腹增强CT结果
实验室检查:
补体C4浓度:0.03g/L ↓(正常值范围:0.1-0.4g/L);C1-INH浓度:0.04g/L ↓(正常值范围:0.21-0.39g/L);C1-INH功能:0.0% ↓(正常值范围:≥68%)。
家系筛查及基因检测:
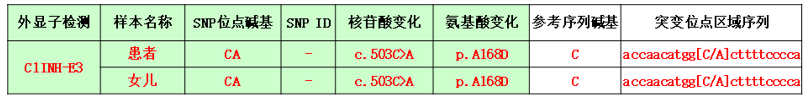
患者及其女儿均存在C1-INH-E3外显子基因突变。
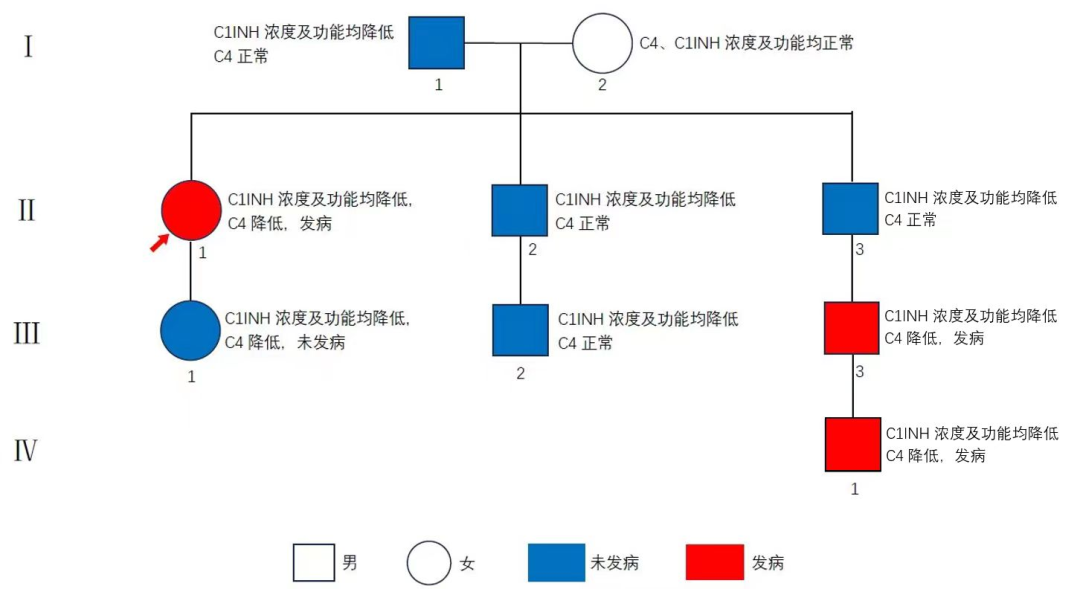
图2 家系检测结果
表1 C1-INH基因检测结果
3 诊断与鉴别诊断
鉴别诊断:
腹痛与急性胃肠炎/阑尾炎、炎症性肠病、缺血性肠病、腹腔动脉压迫综合征、腹型过敏性紫癜、肥大细胞增生症、狼疮肠系膜血管炎、嗜酸粒细胞性胃肠炎等胃肠道疾病及胆囊、胰腺、泌尿系统、妇科疾病进行鉴别诊断。水肿鉴别诊断包括心衰、肝硬化、肾病综合征或其他类型肾病、结缔组织病、药物及过敏因素。
诊断:
患者反复发作腹痛,曾有颜面部、双手及足背部水肿史,水肿具有自限性,全腹增强CT可见十二指肠管壁水肿增厚,且补体C4、C1-INH浓度及功能均显著下降,同时基因检测显示C1酯酶抑制物(C1-INH)基因突变阳性。结合国内外指南共识给出的诊断标准,确诊为遗传性血管性水肿(HAE)。
4 治疗建议
确诊后患者接受拉那利尤单抗注射液(300mg,每2周皮下注射1次)长期预防治疗,治疗后随访半年,未出现腹痛呕吐及皮肤水肿。
思考与讨论
HAE是一种C1-INH浓度和/或功能降低引起的罕见常染色体显性遗传病,患者常反复出现面部、四肢或躯干局限性水肿,且多伴有消化道或喉咽部水肿,继发腹水或呼吸困难,给患者的生命健康带来严重威胁[1]。研究显示,中国七成以上HAE患者存在胃肠道症状,临床表现为腹痛、恶心、呕吐、腹泻和便秘,易被误诊为胃肠炎和阑尾炎,其中24.7%的患者接受了不必要的阑尾切除术或剖腹手术,延误治疗[2]。因此,临床医生需明确如何排查可疑的HAE患者并进行鉴别诊断,进而早期诊断和治疗HAE。
HAE典型影像学特征为急性发作期存在腹水、节段性肠壁/黏膜增厚,但可在自发缓解期完全缓解。对反复发作的腹痛且B超或CT可见肠壁水肿/腹水,且伴有皮肤水肿史、呼吸道水肿史、家族成员有水肿史、抗组胺、糖皮质激素、肾上腺素、奥马珠单抗无效或儿童、青少年时期发病,具有任一情况者应高度怀疑HAE。研究显示,补体C4在HAE患者发作期间筛查的敏感性为81%-96%,因此,2020年HAE管理指南推荐将补体C4水平作为HAE的初始筛查指标[3]。对于高度可疑HAE患者,同时检测补体C4水平和C1-INH浓度和功能,进而明确诊断[4]。
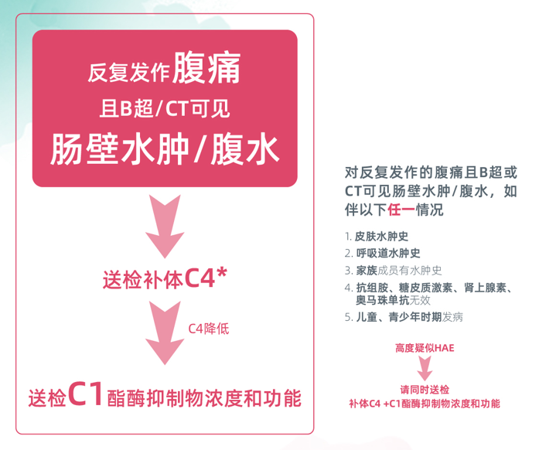
图3 消化科HAE筛查流程图
值得庆幸的是,本例患者入院虽以胃肠道症状为主,但临床医生通过对患者既往病史的了解,给患者进行了全腹增强CT检查,显示十二指肠管壁水肿增厚,鉴别诊断后敏锐察觉到其可能就是HAE,立刻给患者开具了补体C4和C1-INH检查,果不其然,患者的C4和C1-INH水平远低于正常值,与此同时,患者及其女儿接受基因检测后,均发现C1‑INH突变阳性,结合该病人的既往病史,当即诊断其为HAE。
此外,在辅助确诊的同时,基因检测也筛查出了患者女儿这个新的“潜在”HAE患者,这提示临床医生,对HAE患者进行家系基因检测,有助于识别C1‑INH突变基因携带者,特别是无症状或非典型个体,能够帮助他们尽早实现有针对性的预防和治疗,提高患者生活质量,降低病死率[5]。
根据国内外指南推荐,HAE的治疗可分为急性发作的按需治疗、短期和长期预防性治疗,其中,长期预防性治疗的目的是实现疾病的完全控制并使患者生活正常化。拉那利尤单抗可与血浆激肽释放酶结合而抑制其蛋白水解活性,以抑制HAE患者缓激肽生成过多,从而降低水肿发作频次,2021年世界过敏组织/欧洲变态反应与临床免疫学会HAE管理指南[6-7]推荐拉那利尤单抗作为一线长期预防治疗药物。本例患者在确诊后也遵医嘱接受了拉那利尤单抗特异性治疗并取得了良好的治疗效果,随访期间未出现腹痛及皮肤水肿症状。
参考文献:
[1]姚我,汪慧英. 拉那利尤单抗治疗遗传性血管性水肿临床试验的系统综述[J].中华皮肤科杂志,2024, e20230378.
[2]Cao Y, et al. Recurrent and acute abdominal pain as the main clinical manifestation in patients with hereditary angioedema. Allergy and asthma proceedings 2021; 42(2): 131-5.
[3] Busse PJ, et al. US HAEA Medical Advisory Board 2020 Guidelines for the Management of Hereditary Angioedema. J Allergy Clin Immunol Pract. 2021 Jan;9(1):132-150.e3.
[4]周敏,等. 遗传性血管性水肿患者诊疗流程与健康管理模式[J]. 中华预防医学杂志,2023,57(8):1280-1285.
[5]贾薇,等. 一个遗传性血管性水肿家系的临床及基因研究[J]. 中华耳鼻咽喉头颈外科杂志,2022,57(08):980-985.
[6]Maurer M, et al. The international WAO/EAACI guideline for the management of hereditary angioedema-The 2021 revision and update. Allergy. 2022 Jul;77(7):1961-1990.
[7]汪慧英,等. 长期预防性治疗结合按需治疗控制遗传性血管性水肿——2021版WAO/EAACI国际遗传性血管性水肿管理指南解读. 中华预防医学杂志,2024,58(05):698-705.
作者:消化界
版权声明:
本网站所有注明“来源:梅斯医学”或“来源:MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明“来源:梅斯医学”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言







#遗传性血管性水肿# #腹痛#
95