NEJM:完胜Humira!武田Entyvio治疗UC的III期临床进一步结果出炉
2019-09-29 佚名 新浪医药
9月26日,武田制药宣布评估肠道选择性新型抗炎药Entyvio(vedolizumab)治疗溃疡性结肠炎(UC)的III期临床VARSITY(NCT02497469)的进一步结果已发表于《新英格兰医学杂志》。
9月26日,武田制药宣布评估肠道选择性新型抗炎药Entyvio(vedolizumab)治疗溃疡性结肠炎(UC)的III期临床VARSITY(NCT02497469)的进一步结果已发表于《新英格兰医学杂志》。该研究是UC领域首个头对头生物制剂研究,结果显示,在治疗中度至重度活动性UC患者时,与修美乐(Humira)相比,Entyvio在主要终点(第52周临床缓解率)和次要终点(第52周内镜下黏膜愈合率)表现出了优效。
VARSITY是一项随机、双盲、双模拟、多中心、阳性药物对照、IIIb期研究,旨在评估Entyvio静脉注射(IV)与Humira皮下注射(SC)用于中度至重度活动性UC患者治疗一年(52周)的疗效和安全性。该研究共治疗了769例患者(Entyvio治疗组n=383例;Humira治疗组n=386),所有患者在入组前对皮质类固醇、免疫调节剂或除Humira以外的一种肿瘤坏死因子α(TNFα)拮抗剂反应不足、失去反应或不耐受。研究中,25%的患者先前接受过TNFα拮抗剂治疗,609例为抗TNFα初治患者,160例为抗TNFα经治患者。
研究中,患者被随机分为两组:Entyvio IV+安慰剂SC,安慰剂IV+Humira SC。Entyvio治疗组患者在第0、2、6周、之后每8周一次直至第46周接受Entyvio IV 300mg,在第0周、之后每2周一次直至第50周接受安慰剂SC。Humira治疗组在第0周接受Humira SC 160mg、第2周80mg、之后每2周一次40mg直至第50周,同时在第0、2、6周、之后每8周一次直至第46周接受安慰剂IV。研究期间,两个组均不允许剂量增加。主要终点是临床缓解,定义为完整Mayo评分≤2分且无单项>1分。次要终点包括黏膜愈合(定义为Mayo内窥镜单项≤1分)和无糖皮质激素临床缓解(定义为在基线[第0周]时使用口服糖皮质激素的患者已停止口服糖皮质激素,并在第52周实现临床缓解)。
结果显示,研究达到了主要终点,与Humira组相比,Entyvio组在第52周临床缓解率方面表现出优越性(31.3% vs 22.5%,p=0.006)。探索性分析显示,在第52周,与Humira相比,Entyvio在抗TNFα初治患者亚组(34.2% vs 24.3%)和抗TNFα经治患者亚组(20.3% vs 16.0%)取得了更高的临床缓解率。此外,Entyvio组有26.6%的患者在第14周达到临床缓解,Humira组为21.2%。Entyvio组有18.3%的患者实现持久临床缓解,Humira组为11.9%。
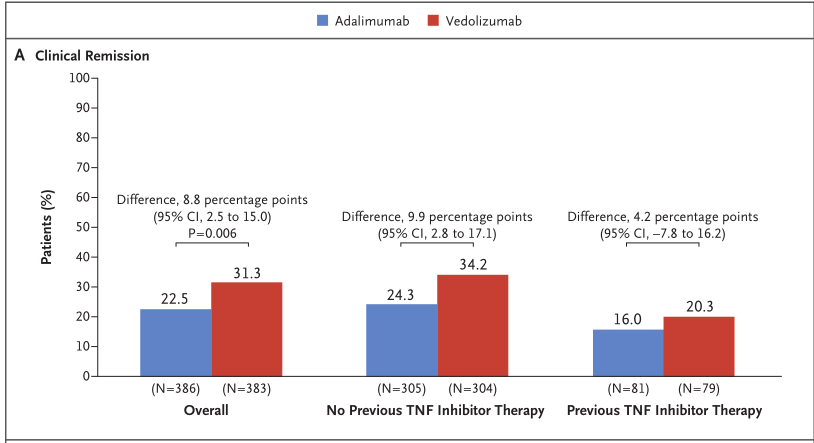
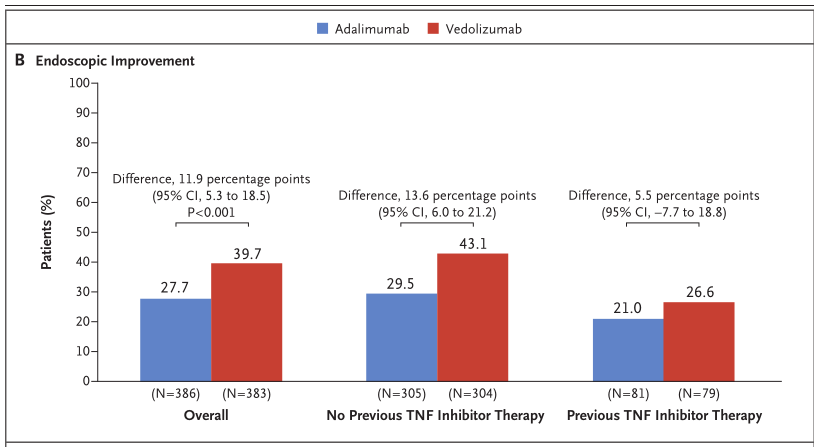
第52周内镜下黏膜愈合率方面,与Humira组相比,Entyvio组显着提高(39.7% vs 27.7%;p<0.001)。
探索性分析显示,在第52周,与Humira相比,Entyvio在抗TNFα初治患者亚组(43.1 vs 29.5%)和抗TNFα经治患者亚组(26.6% vs 21.0%)均取得了更高的内镜下黏膜愈合率。
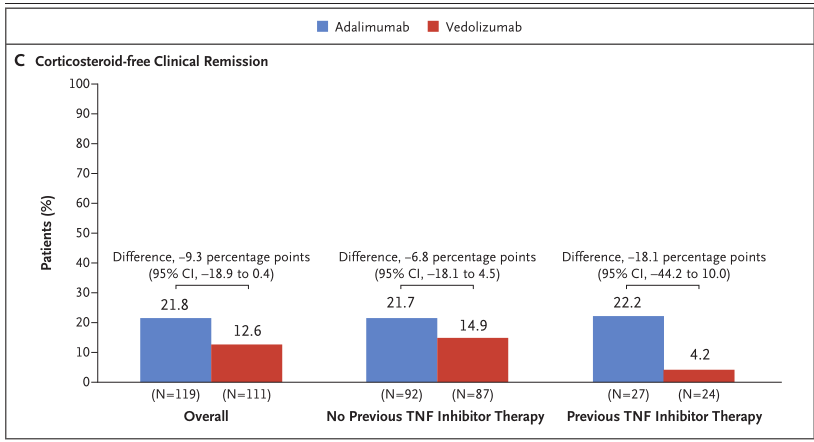
无皮质类固醇临床缓解方面,在基线时使用口服皮质类固醇的患者中,停用皮质类固醇并在第52周取得临床缓解的患者比例Entyvio组低于Humira组(12.6% vs 21.8%),在抗TNFα初治患者亚组(14.9% vs 21.7%)和抗TNFα经治患者亚组(4.2% vs 22.2%)中也低于Humira组。
口服皮质类固醇中位变化的探索性结果显示,从基线至第52周,Entyvio组皮质类固醇使用量中位变化为-10.0mg,Humira组为-7.0mg。
分析结果还表明,Entyvio治疗与生活质量改善相关,Entyvio组有52.0%、Humira组有42.2%在炎症性肠病问卷评分从基线至第52周改善≥16分。问卷检查了炎症性肠病对患者生活四个方面的影响:与原发性肠道疾病直接相关的症状、全身症状、情绪和社会功能。
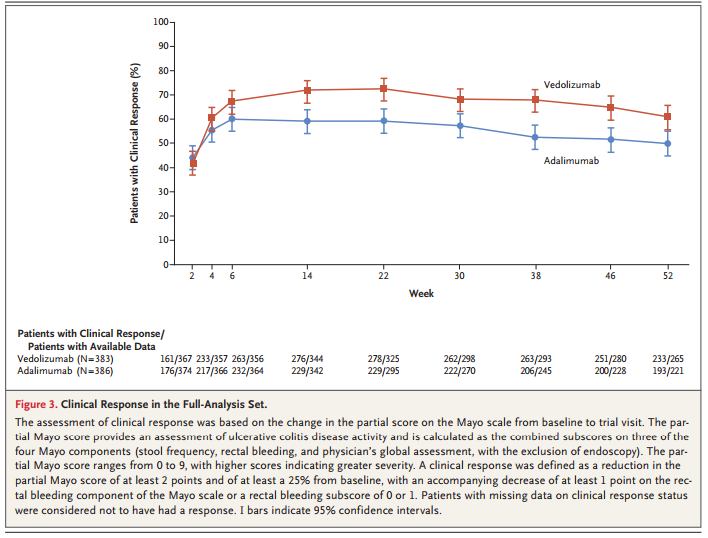
此外,还开展了一项探索性分析,评估Entyvio与Humira对临床反应和无活动性组织学疾病的影响。结果显示,Entyvio组有67.1%在第14周达到临床反应,而Humira组为45.9%,临床反应早在第6周就出现了分离,数据有利于Entyvio。无活动性组织学疾病由GEBOES评分(<3.2)和Robarts病理组织学指数(<5)定义,在第52周,Entyvio组患者达标比例为33.4%和42.3%,Humira组分别为13.7%和25.6%。
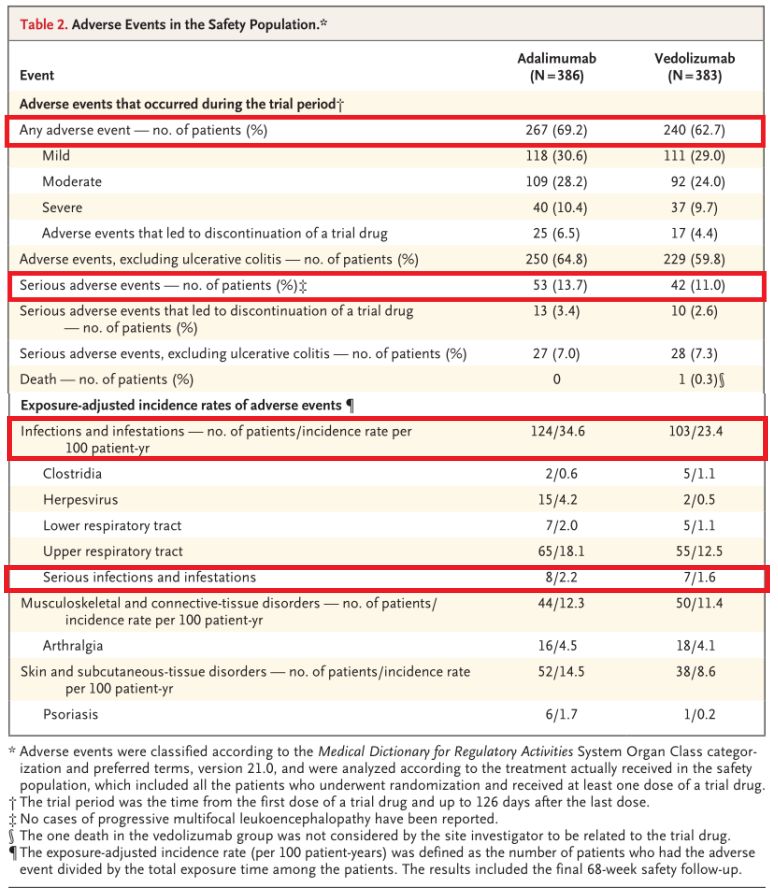
尽管研究并非专门比较两种生物制剂的安全性,但与Humira相比,Entyvio在安全性方面表现更好,具体数据为:在治疗期间,Entyvio组患者的总体不良事件发生率(62.7% vs 69.2%)、严重不良事件发生率(11.0% vs 13.7%)、感染发生率(23.4% vs 34.6%)、严重感染发生率(1.6% vs 2.2%)均较低。
在临床上,治疗UC等慢性致衰性肠道疾病时,使患者在疾病多个方面获得缓解非常重要。VARSITY研究结果为医生提供了宝贵的见解,以支持其在UC患者中启动生物治疗时的治疗决策。
该研究首次在治疗UC方面对两种生物制剂进行了比较,显示了Entyvio与Humira相比在疗效和改善总体生活质量方面的益处。这些数据进一步支持了Entyvio作为一线生物疗法在UC治疗方面的应用。

Entyvio是一种肠道选择性生物制剂,其活性药物成分为vedolizumab,这是一种人源化单克隆抗体,可特异性拮抗α4β7整合素,抑制α4β7整合素对肠道黏膜细胞粘附分子MAdCAM-1的结合。
Entyvio于2014年5月获美国和欧盟批准上市。目前,IV剂型Entyvio已获全球60多个国家/地区批准,用于中度至重度活动性UC或克罗恩病(CD)成人患者的治疗。
原始出处:
Bruce E. Sands et al.Vedolizumab versus Adalimumab for Moderate-to-Severe Ulcerative Colitis.N Engl J Med, September 26, 2019.
作者:佚名
版权声明:
本网站所有注明“来源:梅斯医学”或“来源:MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明“来源:梅斯医学”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言








good
53
#I期临床#
39
#III#
60
#Entyvio#
66
#miR#
45