疑难病例 ǀ AIH、PBC、干燥综合征重叠合并原发性皮肤淀粉样变性1例
2019-08-18 金晶兰 高润平 临床肝胆病杂志
患者女性,48 岁,因“发现肝功能异常6 年,双下肢水肿1 个月余”于2014 年10月28 日入院。患者于2008 年体检时发现肝功能异常,行肝脏穿刺活检,病理诊断为“原发性胆汁性胆管炎- 自身免疫性肝炎(PBC-AIH)重叠综合征”,后口服熊去氧胆酸治疗。2012 年2 月因皮肤色素沉着、口干、眼干就诊于吉林省中日联谊医院,诊断为“干眼症”,同时行右胫前局部皮肤活检,病理诊断为“皮肤淀粉样变”
一、病例介绍
患者女性,48 岁,因“发现肝功能异常6 年,双下肢水肿1 个月余”于2014 年10月28 日入院。患者于2008 年体检时发现肝功能异常,行肝脏穿刺活检,病理诊断为“原发性胆汁性胆管炎- 自身免疫性肝炎(PBC-AIH)重叠综合征”,后口服熊去氧胆酸治疗。2012 年2 月因皮肤色素沉着、口干、眼干就诊于吉林省中日联谊医院,诊断为“干眼症”,同时行右胫前局部皮肤活检,病理诊断为“皮肤淀粉样变”。2012 年5 月就诊于笔者所在医院风湿科,考虑AIH诊断成立,给予甲泼尼龙片6mg/d 联合硫唑嘌呤50mg/d 治疗近3个月,其间多次复查肝功能各指标见好转,后因体态改变停用,规律服用熊去氧胆酸至今(由250mg/d 渐增至1000mg/d),每3 个月复查肝功能一次,提示轻度异常。近1 个月因肝功能异常明显伴双下肢水肿再次入院。患者近2 年皮肤瘙痒,口干、眼干加重,进食哽噎感。否认其他用药史、肝炎及其他肝病家族史,无烟酒史及药物过敏史。
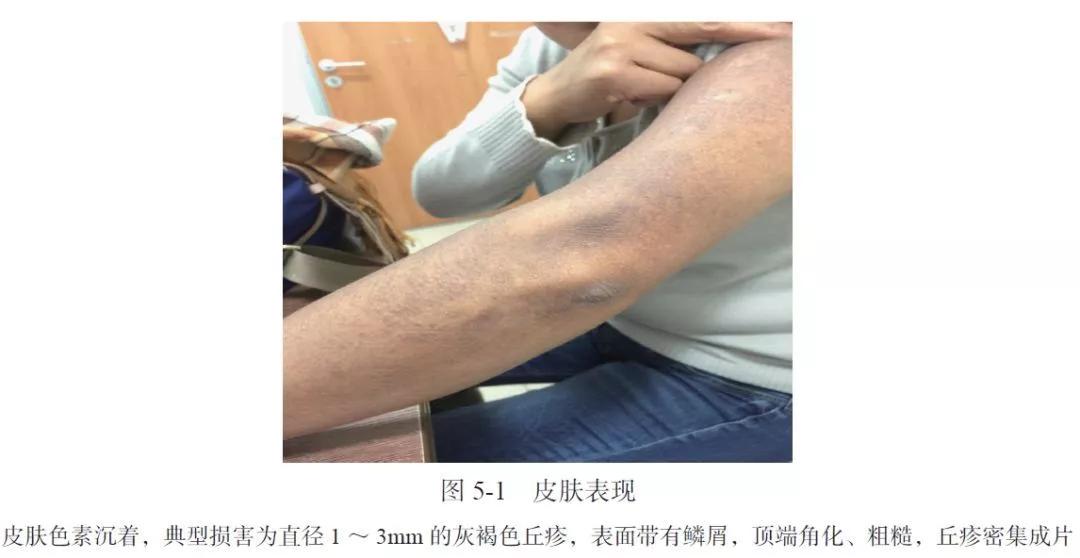
入院查体:皮肤色素沉着,双胫前、上臂、腰背部明显,典型损害为直径1~3mm 的灰褐色丘疹,呈半球形,表面带有鳞屑,顶端角化、粗糙,丘疹密集成片,未见明显融合(图5-1),双下肢轻度水肿,其余未见明显阳性体征。
入院肝功能:AST 102U/L,ALT 71U/L,GGT 459U/L,ALP 340U/L,CHE 3368U/L,Alb 30.4g/L,Glob 49.1g/L,A/G 0.62,TBil78.1μmol/L,DBil 56.4μmol/L,IBil 21.7μmol/L,TG 2.16mmol/L。
免疫三项:IgG 28.60g/L,IgA 5.15g/L,IgM 3.58g/L。ANA 系列:抗干燥综合征A 抗体(抗SSA)阳性,抗核抗体(ANA)颗粒型1∶3200 阳性;抗线粒体M2 抗体(AMA-M 2)>200RU/ml;自身免疫肝病IgG抗体:抗gp210抗体3+。ESR 101mm/h;类风湿因子、抗链球菌溶血素阴性;CRP 6.65mg/L;铁代谢未见异常。
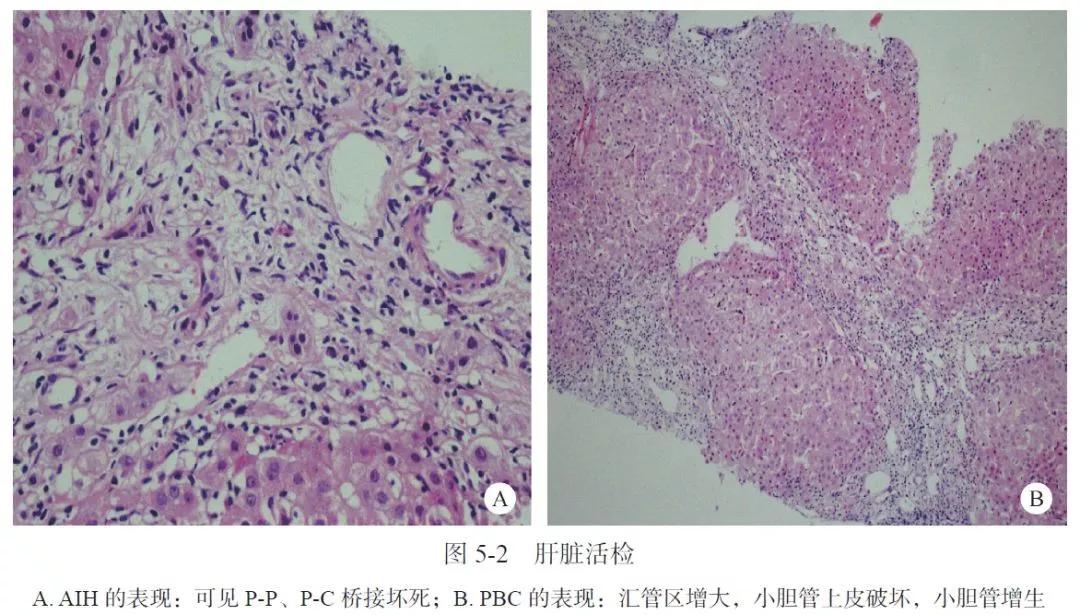
肝脏CT:肝硬化、脾大;考虑门静脉高压伴侧支循环开放(门静脉主干14mm)。2008 年4 月4 日肝脏活检:肝小叶结构基本存在,肝细胞灶状坏死,重度界面炎,可见P-P及P-C桥接坏死,汇管区扩大,炎细胞浸润,小胆管上皮破坏、增生,考虑AIH-PBC重叠综合征(图5-2);
2012年4月6日皮肤活检:皮肤淀粉样变。2014年11月4日笔者所在医院唇腺活检:淋巴细胞浸润,呈灶性分布,浸润灶>1个/4mm2,符合口腔干燥症(图5-3)。

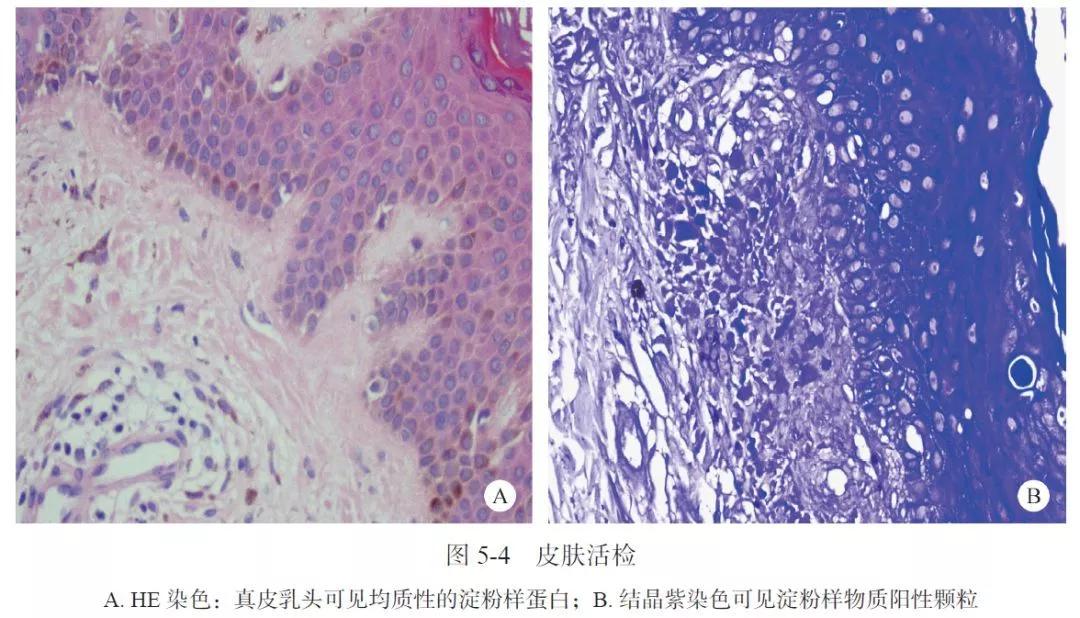
2014年11月5日笔者所在医院皮肤活检:HE染色可见皮肤淀粉样变性(图5-4)。临床诊断:AIH、PBC、干燥综合征三者重叠合并原发性皮肤淀粉样变性,给予甲泼尼龙片8mg/d、硫唑嘌呤50mg/d,熊去氧胆酸750mg/d 治疗1个月后,AST 55U/L,ALT 75U/L,GGT 400U/L,ALP 200U/L,DBil 40μmol/L,ESR 53mm/h,病情明显好转。
二、临床诊治思维过程
AIH-PBC 重叠综合征目前尚无标准化的诊断标准,一般认为PBC或AIH成年患者中有8%~10%可表现为此种变异型。目前,“巴黎标准”常被用于诊断AIH-PBC变异型。但针对每种疾病,至少符合如下两项及两项以上条件方可诊断,诊断AIH:① ALT≥5倍正常值上限(ULN);②血清IgG ≥ 2×ULN 或SMA阳性;③肝活组织检查显示门管区周围或小叶间隔中重度碎屑样坏死伴淋巴细胞浸润。诊断PBC:① ALP≥2×ULN 或GGT≥5×ULN;②抗线粒体抗体阳性;③肝脏活检显示典型的胆管病变。依据上述标准,该患者诊断AIH-PBC重叠综合征是成立的。
原发性皮肤淀粉样变属淀粉样变性病的一型,系指淀粉样蛋白沉积于正常的皮肤组织中、不累及其他器官的一种慢性皮肤病。本病病因尚不明,可能与遗传和免疫有关。临床以苔藓样型和斑疹型皮肤表现较常见。结合该患者皮肤表现及2次皮肤活检组织病理学特征,故可诊断为原发性皮肤淀粉样变。干燥综合征是一种侵犯泪腺、唾液腺等外分泌腺体,以淋巴细胞浸润和特异性自身抗体(抗-SSA/SSB)为特征的弥漫性结缔组织病。临床上主要表现为干燥性角结膜炎、口腔干燥症,还可累及其他多个器官而出现复杂的临床表现。本例患者有明显的眼干、口干表现,既往曾诊断为“干眼症”,此次入院血清抗-SSA 阳性,唇腺病理提示淋巴细胞浸润灶>1 个/4mm2,可确诊为干燥综合征。尽管有干燥综合征合并淀粉样变的报道,但比较罕见,此类患者可发现皮肤、肾脏、乳腺、舌等处淀粉样物质沉积,其中以皮肤受累为主的局限性皮肤结节性淀粉样变合并干燥综合征的发病率为25%。值得注意的是,干燥综合征患者罹患淋巴瘤的概率较正常人高,合并非霍奇金淋巴瘤的发病率为4.3%,对干燥综合征合并淀粉样变患者是否会进展为淋巴瘤,仍需大规模的临床研究。该患者目前未发现系统性淀粉样变性的证据,需继续随访。目前PBC的治疗推荐以熊去氧胆酸(UDCA)为主,AIH治疗以糖皮质激素和其他免疫抑制剂为主,对AIH-PBC重叠综合征的治疗目前还没有成熟的经验。一项系统评价证实,UDCA和糖皮质激素联合应用是AIH-PBC重叠综合征的最佳治疗选择。该患者所患上述4种疾病均为激素及免疫抑制剂治疗适应证,因此在本次住院开始给予糖皮质激素和硫唑嘌呤联合治疗,同时予以UDCA治疗,使临床症状、肝功能生化及组织学改变均有所改善。但患者肝硬化已经发展至肝功能失代偿期,预后差。
三、诊疗体会
AIH-PBC与干燥综合征三者重叠合并原发性皮肤淀粉样变性目前尚无报道,但考虑到这种疾病病情复杂,诊断相对困难。因此,临床医生要及时发现、早期明确诊断,并给予合理治疗。通过对该患者6 年的病史分析,发现其临床表现序贯出现,自身免疫性疾病持续进展,由于AIH、PBC 和干燥综合征三者均属于自身免疫性疾病,它们与原发性皮肤硬化症是否存在相关性,仍需进一步研究。因此在治疗上仍需要综合考虑,既有共性免疫抑制治疗(糖皮质激素和硫唑嘌呤联合),也有针对PBC的个性治疗(UDCA治疗)。
四、专家点评
这是关于“AIH、PBC、干燥综合征三者重叠合并原发性皮肤淀粉样变性”的1例病例报告。该患者集多种疾病于一身,数年间几乎同时发病,在多家医院同时就诊、同时治疗,最终确诊,实属少见。病例报告中对患者各种疾病的症状体征、就诊经过、检查结果及治疗过程描述清楚,诊断明确,尤其难能可贵的是还提供了多种组织病理活检图片等相关资料,包括肝脏、唇腺、皮肤等,是一份很好的病例报告,有重要的临床参考和借鉴价值。当然,该病例中涉及的具体病种并不是罕见病。干燥综合征(SS)是一种侵犯外分泌腺体,尤以侵犯唾液腺和泪腺为主的慢性自身免疫性疾病。主要表现为口、眼干燥,也可有多器官、多系统损害。受累器官中有大量淋巴细胞浸润,血清中多种自身抗体阳性。常与其他风湿病或自身免疫性疾病重叠。因此,干燥综合征和AIH-PBC重叠综合征共同存在的病例临床并不罕见,或者说这些疾病其实是一种疾病的不同器官或系统受损的各自表现。因为干燥综合征患者多因症状轻微不就诊,或仅在口腔科、眼科等专科就诊,所以肝病专科较少见到这类患者,而AIH、PBC或二者重叠综合征患者则不少见,提示肝病专科医生在诊断AIH、PBC时应关注是否合并有干燥综合征,应做相应的辅助检查。自身免疫性疾病的主要治疗用药是激素或联合免疫抑制剂,需要早期和长期用药。单纯PBC早期用熊去氧胆酸治疗效果良好,可以阻止病情进展。一旦发展为肝硬化,伴有胆红素升高,则疗效有限。该患者就是如此,虽经积极治疗,仍有多项指标异常。原发性皮肤淀粉样变又称苔藓样和斑疹型淀粉样变;属淀粉样变性病的一型,系指淀粉样蛋白沉积于正常的皮肤组织中而不累及其他器官的一种慢性皮肤病。临床并不少见,呈慢性经过,迁延多年,可自行消退,但仍可复发。该患者的自身免疫性疾病合并原发性皮肤淀粉样变可能纯属巧合,并不存在必然的相关性。
作者:金晶兰 高润平
版权声明:
本网站所有注明“来源:梅斯医学”或“来源:MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明“来源:梅斯医学”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言
小提示:本篇资讯需要登录阅读,点击跳转登录



#重叠合并#
43
#PBC#
44
干燥综合征的治疗。
92
#AIH#
49
#疑难病例#
39