
JCI Insight.:GCN2 激酶激活介导肺血管重塑和肺动脉高压
2024-10-05 刘少飞 MedSci原创
研究发现,Eif2ak4基因缺失的小鼠(KO小鼠)未表现出自发性肺静脉阻塞病(PVOD)和肺动脉高压(PH)。然而,GCN2激酶结构域的缺失减轻了缺氧诱导的肺血管重塑和PH。

肺动脉高压(PH)的定义是静息平均肺动脉压力超过20 mmHg。尽管PH有多种病因,其发展几乎总是伴随症状加重和死亡率增加。根据病理生理机制、临床表现、血流动力学特征和治疗管理,PH被临床分为五组。肺动脉高压(PAH,第1组)的特点是肺血管重塑和肺血管阻力逐渐增加,最终导致右心室肥厚和右心衰竭。近年来,PAH治疗取得了一定进展,显著改善了患者预后。然而,目前批准的PAH药物主要集中在减少肺血管收缩,仅提供部分症状缓解。由于对导致肺血管细胞功能障碍的分子机制了解不足,缺乏直接针对肺血管重塑的治疗,导致PAH的五年死亡率仍高达50%。GCN2是一种广泛存在于真核生物中的丝氨酸-苏氨酸激酶,主要作为代谢应激的传感器。在氨基酸饥饿的情况下,GCN2通过未充足的tRNA与其相关域结合而激活,随后抑制一般蛋白合成并选择性激活应激蛋白合成。近年来研究还发现,GCN2能响应紫外线照射、氧化应激和缺氧等多种其他应激。除了调节翻译起始,GCN2还与G1期停滞、细胞凋亡、肿瘤生长和炎症等过程相关。
GCN2(通用调控非抑制激酶2)是一种广泛存在于真核生物中的丝氨酸-苏氨酸激酶,主要作为代谢应激的传感器,例如氨基酸、葡萄糖或嘌呤的限制。当细胞经历氨基酸饥饿时,未充足的tRNA与GCN2的组氨酸-tRNA合成酶相关域结合,激活GCN2,引发构象变化和自磷酸化。随之,GCN2磷酸化真核翻译起始因子2(EIF2α),抑制一般蛋白质合成,同时选择性激活应激蛋白质合成,以适应氨基酸饥饿。GCN2的激活需要在其激活环的磷酸化,尤其是酵母GCN2的Thr887位点(小鼠的Thr898和人类的Thr899)。这一磷酸化锁定激酶域为开放的活性状态,使其能够在没有tRNA的情况下结合底物,如EIF2α。除了对营养不足的响应,近期研究还表明,GCN2能被紫外线照射、氧化应激和缺氧等多种其他应激激活。除了在调节翻译起始中的作用,GCN2还与G1期停滞、细胞凋亡、肿瘤生长及炎症等过程相关。这些发现突显了GCN2在细胞应激反应和代谢适应中的关键角色,为理解其在多种疾病中的作用提供了重要线索。
EIF2AK4基因(编码GCN2)的隐性突变及其导致的GCN2蛋白表达减少与遗传性肺静脉阻塞病(PVOD)相关,这是一种罕见的严重肺动脉高压(PAH)亚型,特征为隔膜静脉和前隔膜小静脉的内膜增生和纤维化。PVOD亚组的预后较经典PAH差,目前除了肺移植外没有有效治疗。双等位基因EIF2AK4突变在25%的组织学确认的散发PVOD病例中发现。虽然在特发性PAH(IPAH)患者中EIF2AK4突变较少见,但在一些遗传性PAH患者中也有发现。我们研究了Eif2ak4-/-(KO)小鼠是否会自发发展为PVOD及GCN2信号在肺血管重塑和PAH发展中的作用。令人惊讶的是,KO小鼠并未发展为自发性PVOD和肺动脉高压(PH)。在慢性缺氧刺激下,KO小鼠的PH反而减轻。将内皮靶向的纳米颗粒递送Edn1质粒DNA以恢复Gcn2缺乏小鼠内皮细胞中的Edn1表达,部分逆转了缺氧引起的PH减少表型。此外,GCN2激酶抑制剂A-92处理也抑制了单氟烯诱导的PH。在IPAH患者中,我们观察到GCN2在Thr 899处的显著磷酸化,即GCN2在肺血管内皮细胞中的激活。我们的研究表明,GCN2激酶激活在促进肺血管重塑和PAH发展中具有不利作用,因此针对GCN2激酶信号的治疗可能是针对没有EIF2AK4隐性突变的PAH患者的一个有前景的治疗方法。
研究发现,为了确定 KO 小鼠是否发生自发性 PVOD 或 PH,该研究进行了血流动力学测量和肺静脉闭塞组织学评估。意外的是,Eif2ak4-/-小鼠中GCN2激酶活性的缺失并未诱导PVOD或PH,但抑制了缺氧引起的PH。
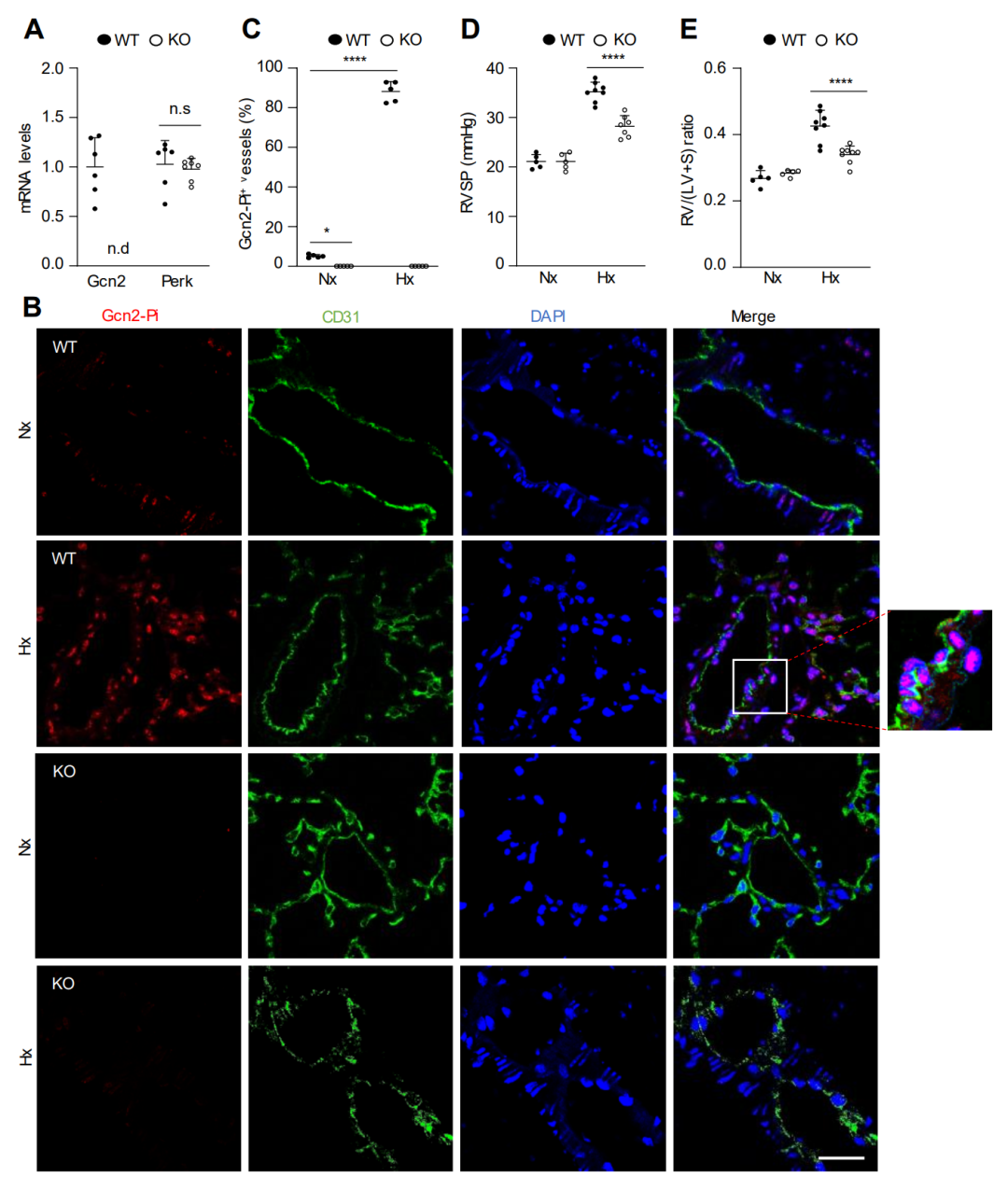
图 1. GCN2 缺乏减轻了小鼠缺氧诱导的 PH
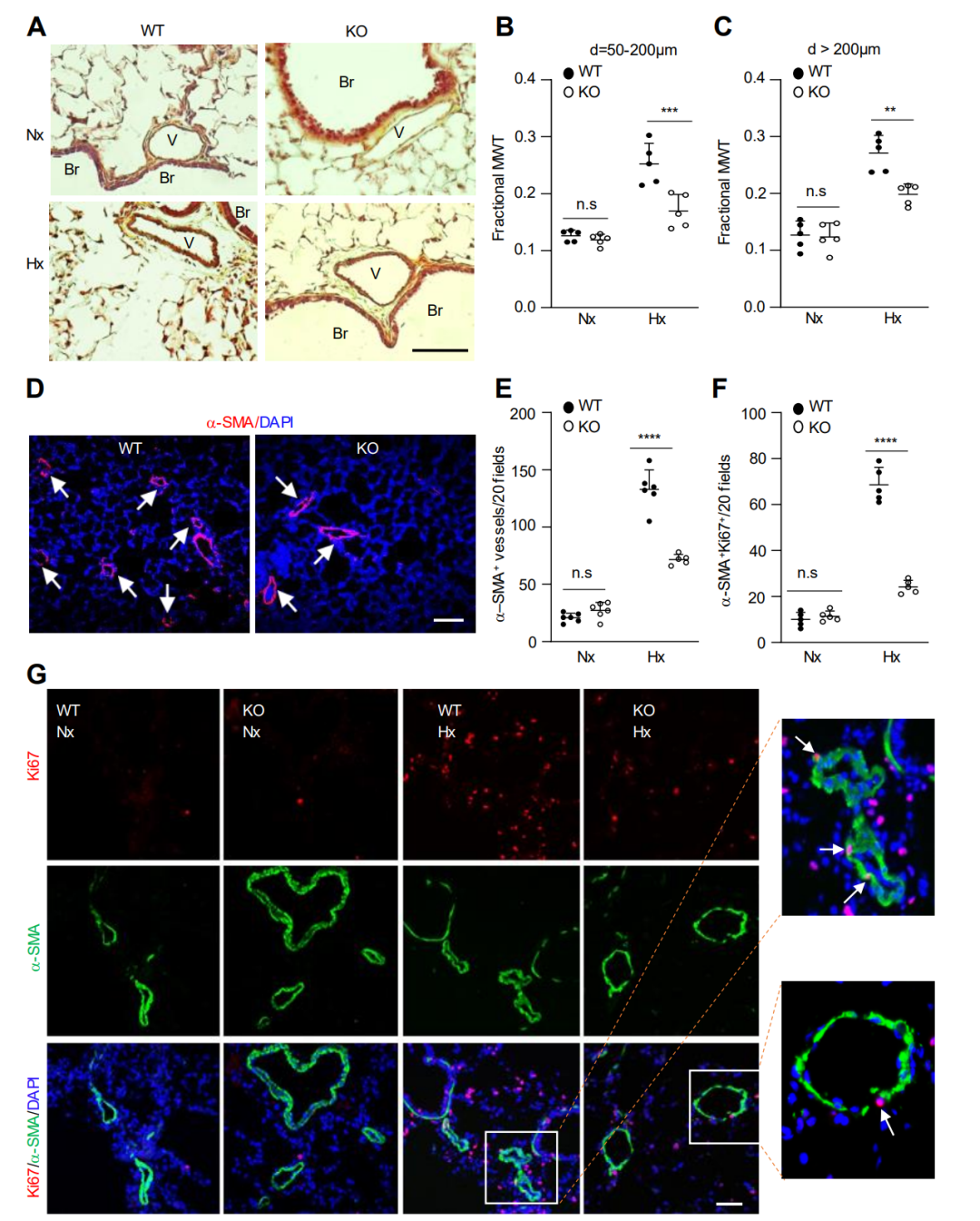
图 2. 与缺氧WT小鼠相比,缺氧KO小鼠的肺血管重塑减少
为了确定 GCN2 是否在体外对缺氧作出反应而被激活,我们用缺氧(1% O2)对 HLMVEC 的原代培养物进行处理。
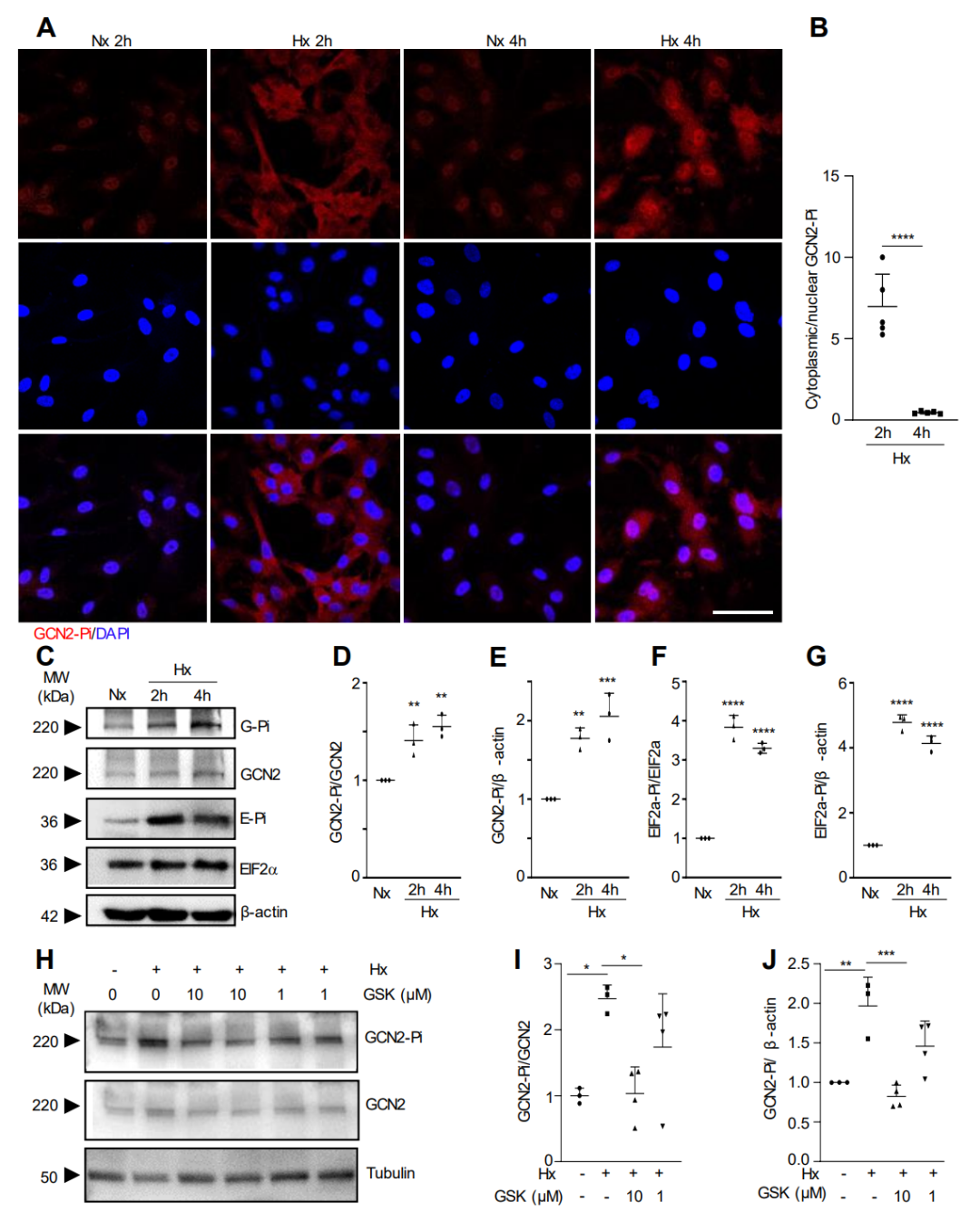
图3.缺氧诱导GCN2磷酸化和活化
为了确定GCN2在调节PH和血管重塑中的分子机制,我们对处于常氧和缺氧条件下的WT和KO小鼠肺组织进行了全转录组RNA测序分析。RNA测序分析表明内皮素-1(Edn1)是GCN2的下游靶标。GCN2通过HIF-2α介导人肺内皮细胞中缺氧引起的Edn1表达。
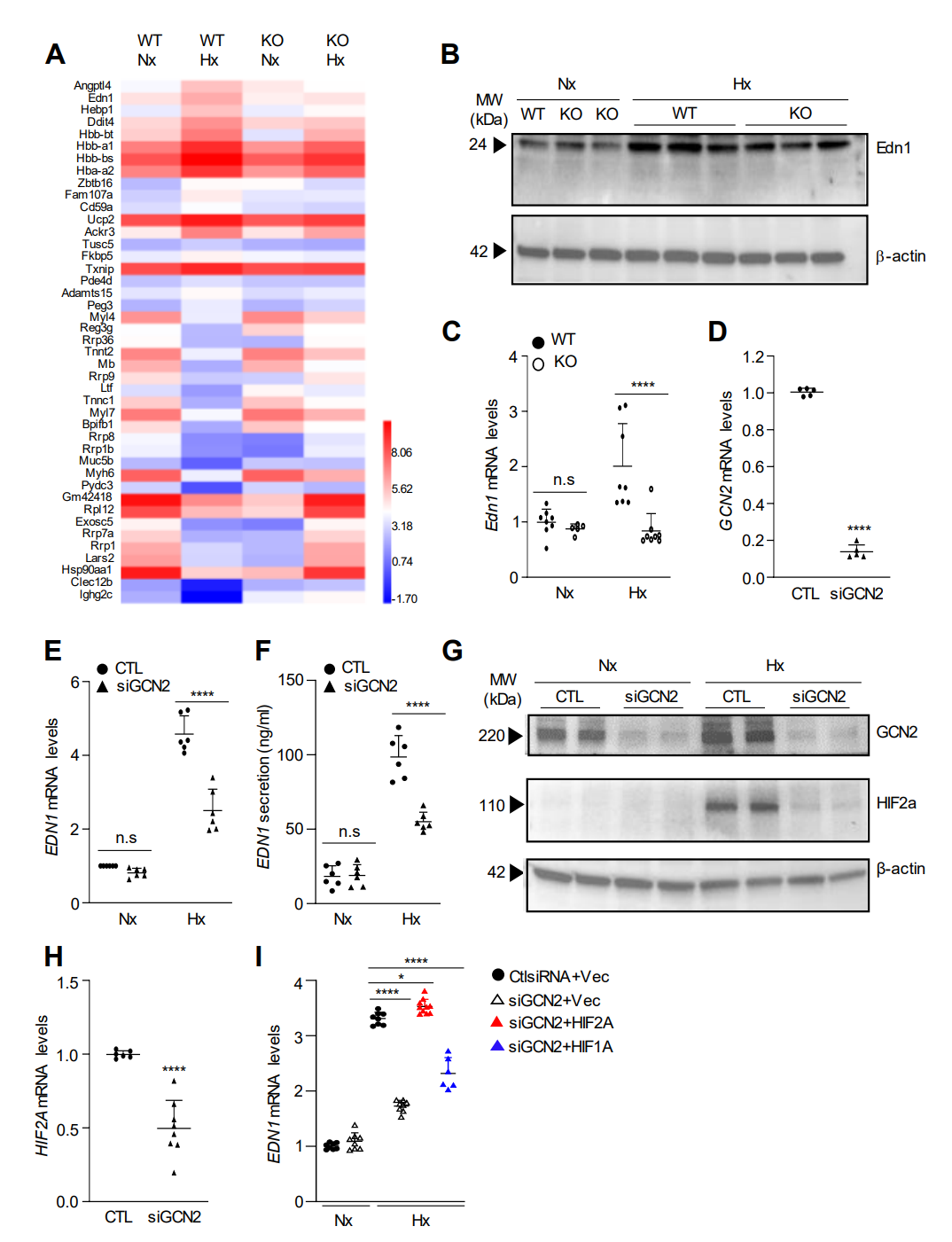
图 4. 缺氧通过GCN2诱导小鼠肺和人肺EC中的Edn1表达
研究接下来进行了体内研究,采用内皮靶向纳米粒子递送基因,以确定KO 小鼠中内皮 Edn1 表达的恢复是否可以逆转因缺氧而降低的PH表型。
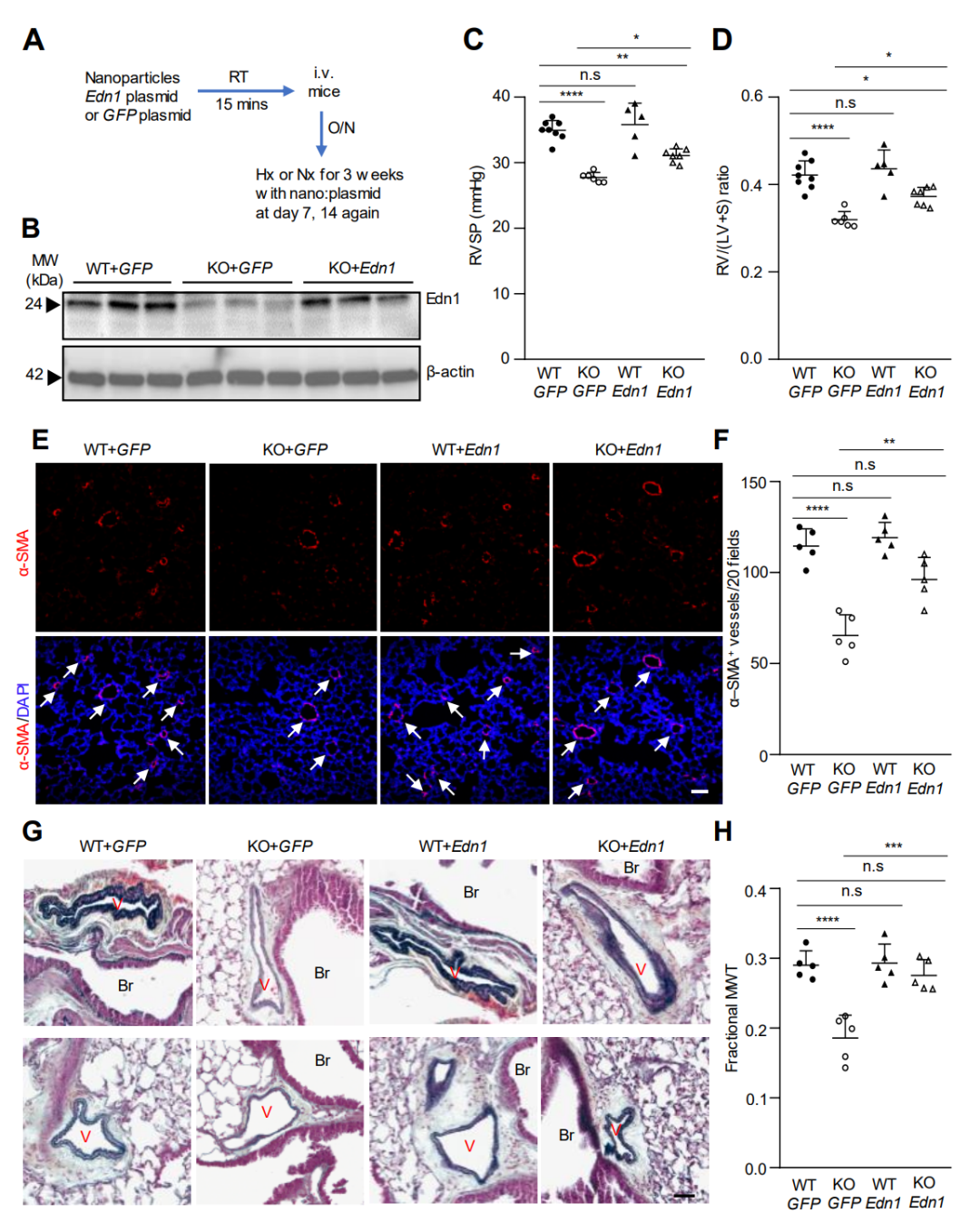
图 5. Gcn2缺陷小鼠中内皮细胞Edn1表达恢复可部分逆转降低的PH表型
恢复Eif2ak4-/-小鼠内皮细胞中Edn1的表达部分逆转了缺氧引起的PH的减少表型。此外,GCN2激酶抑制剂A-92的处理减轻了MCT处理大鼠的PAH。这些研究表明,GCN2激酶的激活在肺血管重塑和PAH中起到介导作用,至少部分是通过Edn1。因此,针对GCN2激酶激活的治疗策略是治疗没有EIF2AK4功能丧失突变的PAH患者的一个有前景的策略。
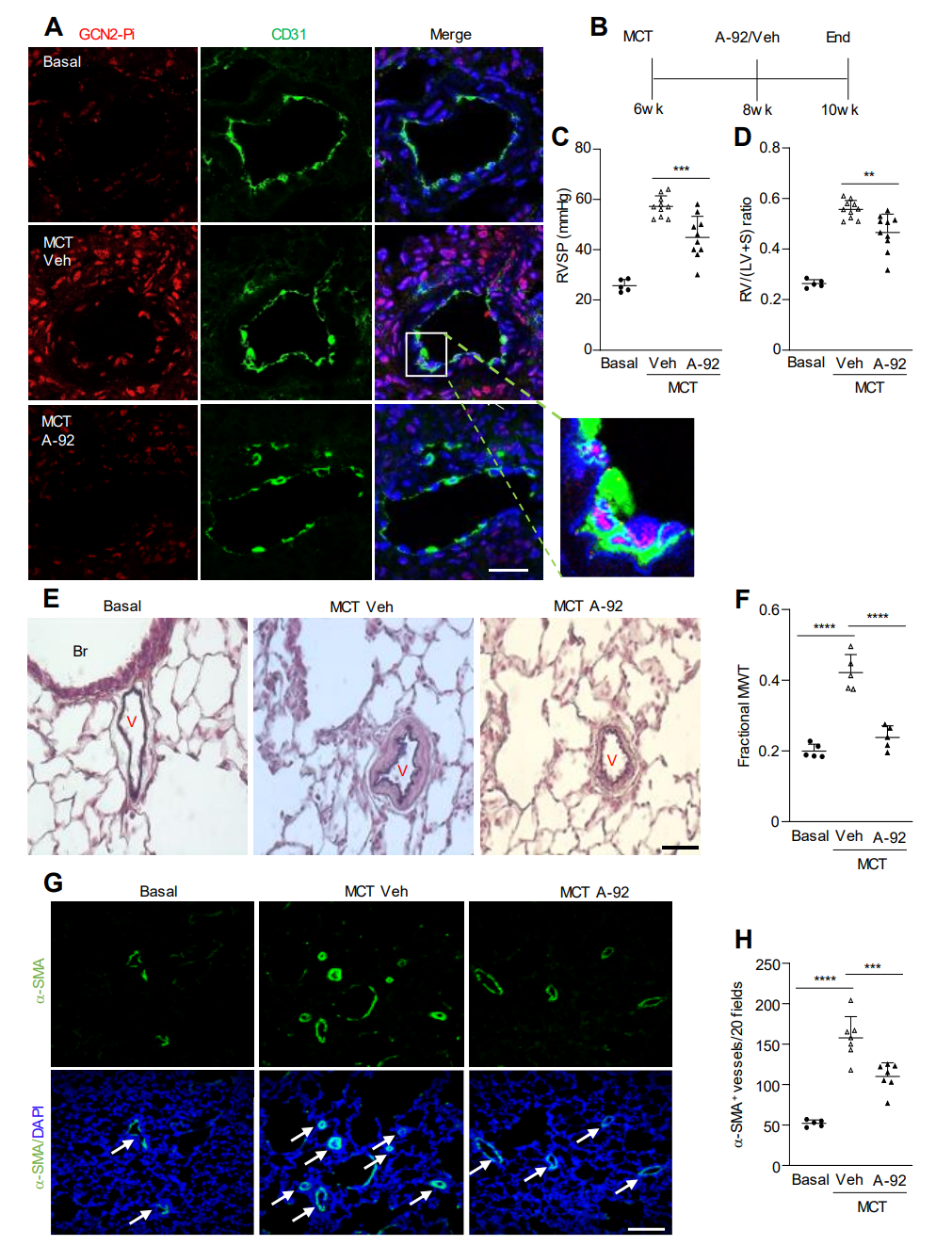
图 6. 药理学抑制GCN2激酶可减轻大鼠中MCT诱发的PAH
最后,在缺氧小鼠、野百合碱处理的大鼠和PAH患者的肺血管内皮细胞中,GCN2被超磷酸化并激活。
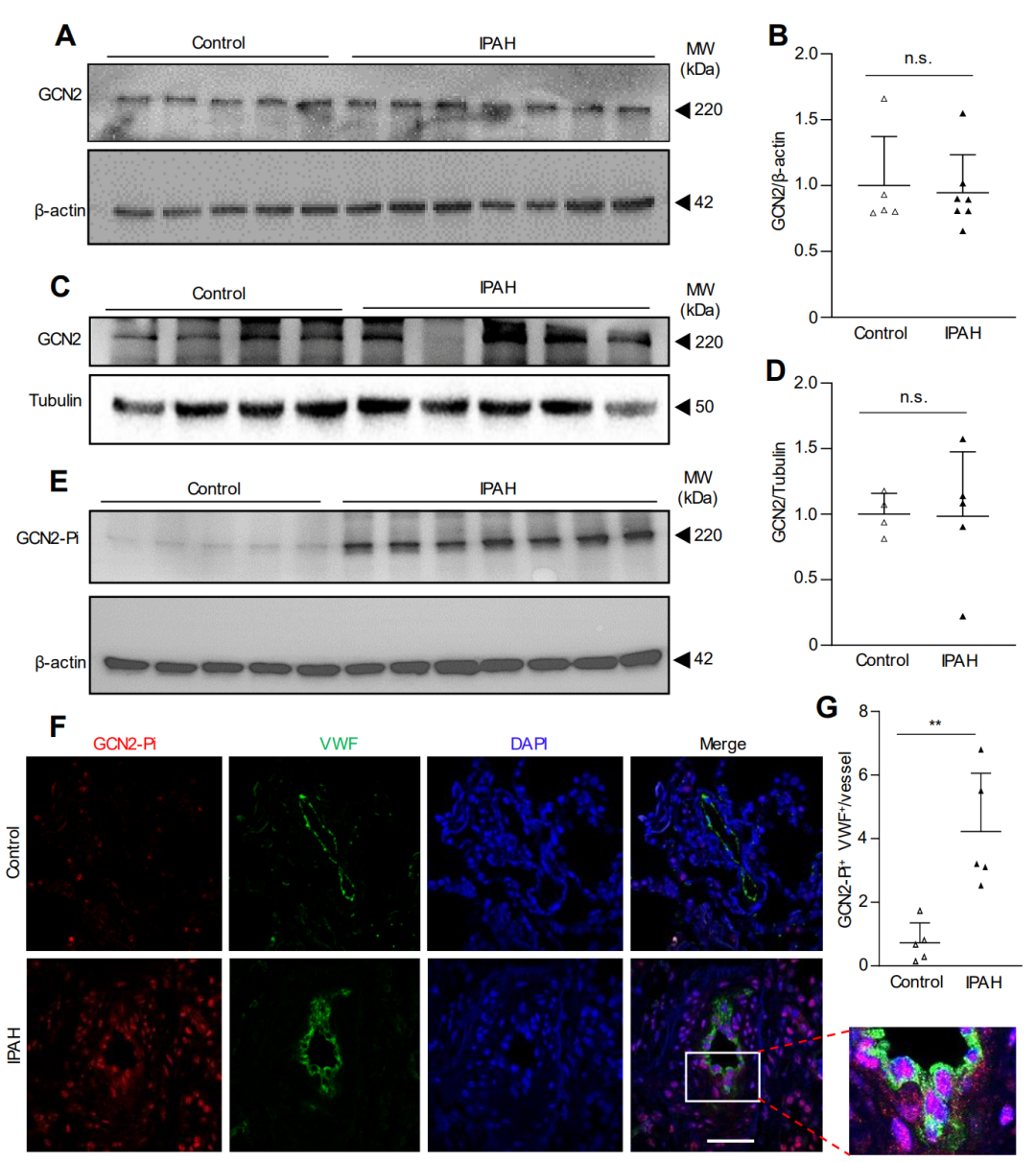
图 7. IPAH 患者肺血管病变 EC 中显著的 GCN2 磷酸化/活化
总之,本研究支持GCN2激酶激活在肺血管重塑和PAH发展中的重要作用。研究发现,Eif2ak4基因缺失的小鼠(KO小鼠)未表现出自发性肺静脉阻塞病(PVOD)和肺动脉高压(PH)。然而,GCN2激酶结构域的缺失减轻了缺氧诱导的肺血管重塑和PH。
机制研究表明,GCN2在正常人肺微血管内皮细胞(HLMVECs)中的激活通过HIF-2α介导缺氧诱导的内皮素-1(EDN1)表达。在缺氧刺激下,恢复小鼠肺内皮细胞中的Edn1表达部分逆转了KO小鼠的PH减轻表型。此外,GCN2激酶活性的抑制(通过A-92处理)同样减轻了单氟烯诱导的PAH及肺血管重塑。
在特发性PAH(IPAH)患者的肺组织中,GCN2的磷酸化水平在肺血管内皮细胞中显著增加,但GCN2的表达变化不大。综上所述,这些数据表明GCN2激酶激活在肺血管重塑和PAH发展中起着必不可少的作用。因此,针对GCN2信号的治疗策略可能为没有GCN2功能丧失突变的PAH患者提供新的治疗思路。
原始出处:
Zhu MM, Dai J, Dai Z, Peng Y, Zhao YY. GCN2 kinase activation mediates pulmonary vascular remodeling and pulmonary arterial hypertension. JCI Insight. 2024 Sep 24:e177926. doi: 10.1172/jci.insight.177926. Epub ahead of print. PMID: 39316438.
作者:刘少飞
版权声明:
本网站所有注明“来源:梅斯医学”或“来源:MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明“来源:梅斯医学”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言







#肺动脉高压# #肺血管重塑# #GCN2 #
86