
【病例报告】HTRA丝氨酸肽酶1基因杂合突变相关脑小血管病一例
2024-08-10 中国脑血管病杂志 中国脑血管病杂志
本研究总结1例HTRA1基因杂合突变相关CSVD患者的临床资料、影像学表现,以期为神经科医师对该病的认识及临床诊疗提供参考。
摘要:报道1例诊断为HTRA丝氨酸肽酶1(HTRA1)基因杂合突变相关脑小血管病(CSVD)的52岁女性患者。该例患者既往无高血压病、糖尿病史,无烟酒嗜好;其外祖父、外祖母为近亲结婚,外祖母及母亲死于脑梗死;临床表现为复发性脑梗死、轻度认知障碍,头部MRI示多发腔隙性脑梗死、广泛脑白质变性和微出血病灶;全外显子组基因检测报告示HTRA1c.947A>G杂合突变。对于CSVD患者应追问其家族史,对疑似遗传性CSVD患者,需考虑存在HTRA1基因杂合突变的可能;并合理借助基因检测方法,筛选CSVD高危家族患者并进一步指导治疗。
脑小血管病(cerebral small vessel disease, CSVD)是指各种因素影响脑内小血管所导致的一组异质性疾病,是卒中和血管性认知障碍的主要原因之一。随着基因检测技术的发展,越来越多与遗传相关的CSVD被报道,以常染色体显性遗传性脑动脉病伴皮质下梗死和白质脑病(cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy,CADASIL)最常见,而HTRA丝氨酸肽酶1(high-temperature requirement A serine peptidase 1, HTRA1)基因纯合突变导致的常染色体隐性遗传性脑动脉病伴皮质下梗死和白质脑病(cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy, CARASIL)多来自于近亲家庭。有研究报道,HTRA1基因杂合突变也可致常染色体显性遗传性CSVD。目前,已有50余个HTRA1基因杂合突变位点被报道。本研究总结1例HTRA1基因杂合突变相关CSVD患者的临床资料、影像学表现,以期为神经科医师对该病的认识及临床诊疗提供参考。
患者
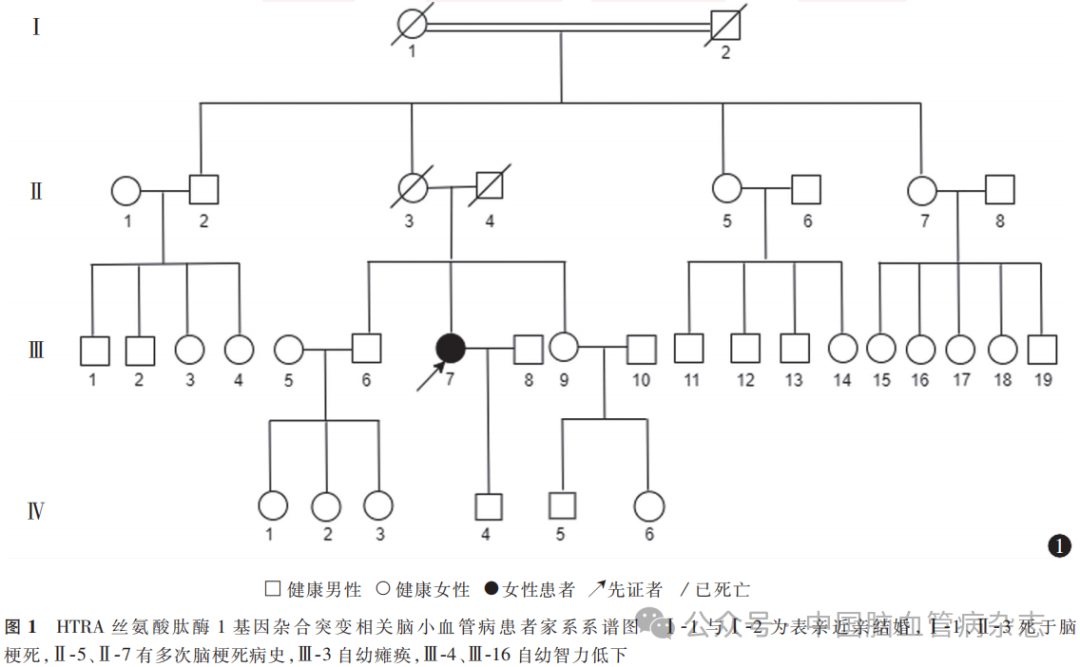
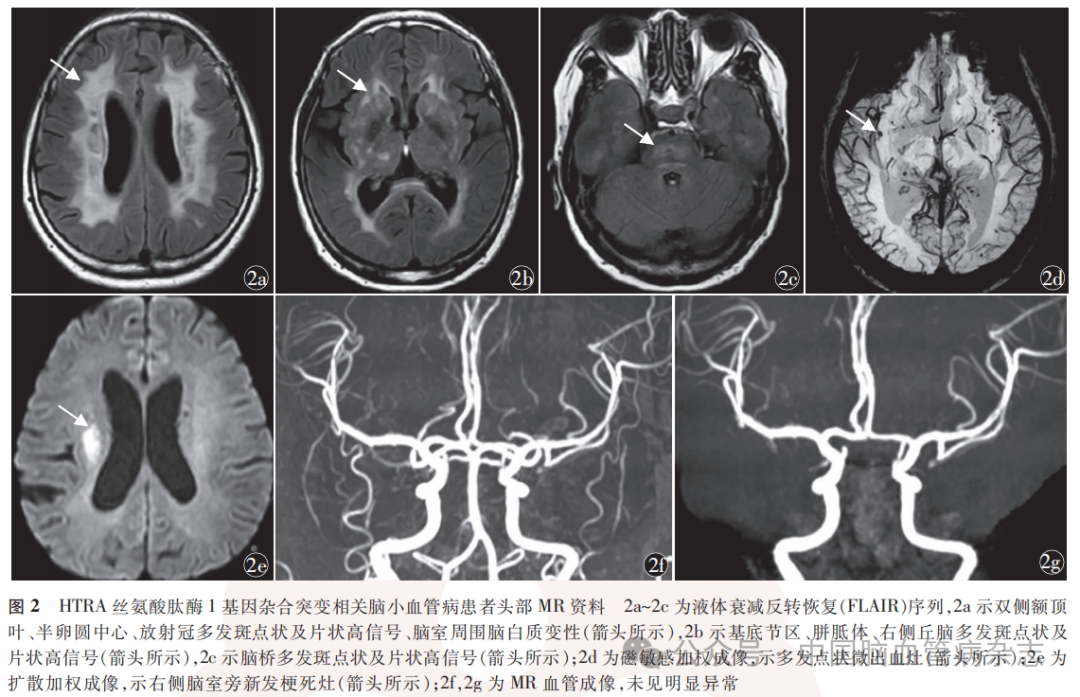
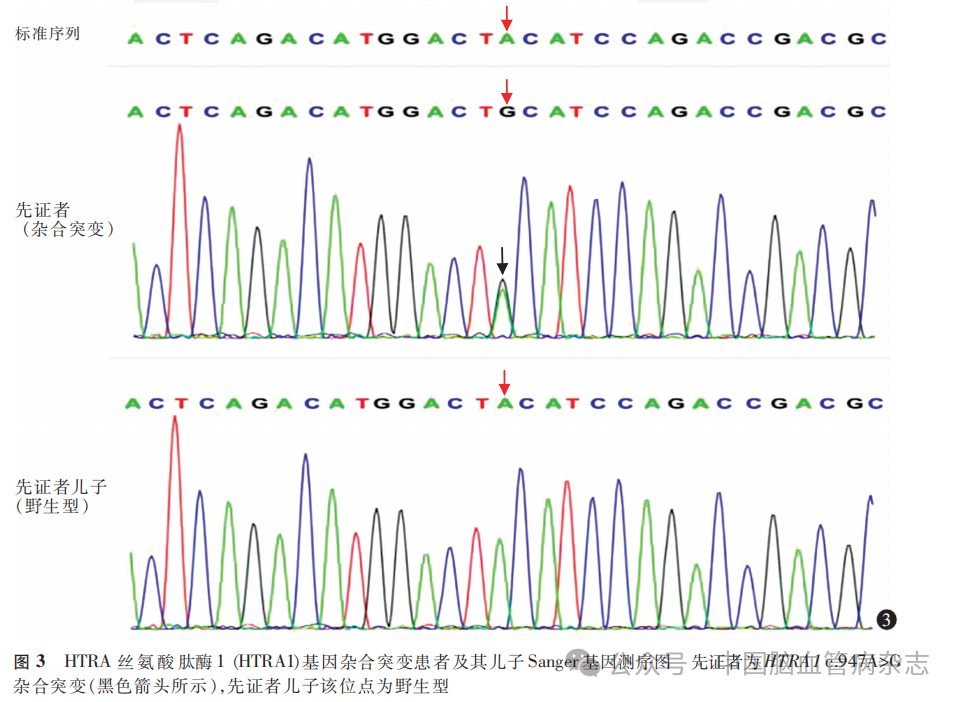
女,52岁,因“间断左手麻木2d,加重伴左下肢无力1d”于2023年8月23日入住河北医科大学第一医院神经内科治疗。住院前2d,患者无明显诱因出现左手发作性麻木,约半小时可自行缓解,左上肢、下肢均正常。住院前1d患者症状加重,左手持续性麻木,伴左下肢无力,行走向左侧歪斜,无上肢乏力,无视物模糊、重影,无头晕、恶心、呕吐,遂就诊于河北医科大学第一医院神经内科,以“脑梗死”收住院。既往史:1年前经当地医院诊断为脑梗死,给予抗血小板聚集、降脂、稳定斑块、改善循环等治疗,遗留左侧口角下垂、饮水呛咳、左侧肢体欠灵活、反应迟钝、记忆力减退。无高血压病、糖尿病史,无烟酒嗜好。家族史:其外祖父、外祖母为表兄妹近亲结婚,外祖母、母亲均死于脑梗死;患者诉其两位姨妈有多次脑梗死病史,其两位表妹自幼智力低下,1位表姐自幼瘫痪。孕1产1,儿子身体健康。图1为家系系谱图。入院体格检查:血压124/80mmHg,无脊柱弯曲、脱发等,心肺腹未见异常。记忆力下降,计算力减退(86-7=?),定向力、理解力正常。左侧口角下垂。左下肢肌力Ⅳ级,左上肢远端肌力Ⅴ-级、近端Ⅴ级,余肢体肌力Ⅴ级;左上肢针刺觉减退,左侧共济运动欠稳准;左侧巴宾斯基征(+),右侧巴宾斯基征(-)。余神经系统体格检查未见异常。实验室检查(2023年8月24日):高密度脂蛋白1.74mmol/L(参考范围1.04~1.55mmol/L),脂蛋白α421mmol/L(参考范围10~140mmol/L),狼疮抗凝物S25.4s(参考范围29.0~49.0s),狼疮抗凝物二氧化硅凝固时间法标准化比值0.55(参考范围0.84~1.16),狼疮抗凝物稀释蝰蛇毒时间法21.4s(参考范围27.0~42.0s),狼疮抗凝物稀释蝰蛇毒时间法确证试验22.1s(参考范围24.0~31.0s),狼疮抗凝物稀释蝰蛇毒时间法标准化比值0.87(参考范围0.92~1.11),余未见异常。头部MR(2023年8月24日):液体衰减反转恢复(FLAIR)序列示双侧额顶叶、半卵圆中心、放射冠多发斑点状及片状高信号、脑室周围脑白质变性(图2a),基底节区、胼胝体、右侧丘脑多发斑点状及片状高信号(图2b),脑桥多发斑点状及片状高信号(图2c);磁敏感加权成像(susceptibility-weighted imaging,SWI)示多发点状微出血灶(图2d);扩散加权成像(DWI)示右侧脑室旁新发梗死灶(图2e);MR血管成像(MRA)未见明显异常(图2f,2g)。初步诊断:脑梗死(小动脉闭塞型)。2023年8月26日,简易精神状态检查(mini-mental state examination,MMSE)量表评分25分,蒙特利尔认知评估(Montreal cognitive assessment,MoCA)量表评分21分。心电图、心脏彩色多普勒超声、颈部血管彩色多普勒超声等相关检查均未见异常。结合临床表现、头部MR特点及MoCA评分结果,考虑为CSVD,给予抗血小板聚集、降脂、稳定斑块、改善循环、改善认知功能等治疗。由于患者家族中有近亲结婚、脑梗死病史,考虑可能存在CSVD家族史,拟对患者进行全外显子组基因检测。征得患者同意并签署知情同意书后,于2023年8月28日采集患者及其儿子的外周血各3ml,通过芯片捕获高通量测序的方法进行全外显子组基因检测。经聚合酶链反应扩增后,Sanger测序验证全外显子组基因检测结果(2023年9月20日):患者存在HTRA1c.947A>G杂合突变,该突变导致第316位的酪氨酸突变为半胱氨酸,其子为野生型(图3)。使用ChimeraX网站(https:// www.novopro.cn/tools/protein - hydrophilicity - plot.html)进行HTRA1c.947A>G杂合突变蛋白结构预测,显示该突变基因第316位氨基酸由酪氨酸突变为半胱氨酸,其氢键未发生改变,其亲水值由1.2变为0.7,突变后亲水性变弱。最终诊断:遗传性CSVD(HTRA1c.947A>G杂合突变),脑梗死(小动脉闭塞型)。患者共住院8d,于2023年8月30日好转出院,嘱其继续口服氯吡格雷、瑞舒伐他汀、多奈哌齐等药物。已随访近10个月,未复发缺血性卒中。
讨论
本例患者为急性起病,以肢体麻木、无力为主诉,既往脑梗死病史,神经系统体格检查示左侧肢体肌力下降、痛觉减退、共济运动欠稳准、巴宾斯基征(+),头部MRI示多发脑梗死病灶,初步诊断为脑梗死(小动脉闭塞型)。因MoCA量表评分21分,认知功能下降,头部MRI示右侧脑室旁新发梗死灶、广泛脑白质变性、脑微出血,故考虑CSVD的诊断。由于患者家族中有近亲婚配,追问家族史提示先证者的外祖母、母亲、两位姨妈均有脑梗死病史,存在可疑CSVD家族史,随后对患者及其儿子进行全外显子组基因检测,结果显示,患者存在HTRA1c.947A>G杂合突变。
HTRA1基因突变与多种疾病相关,如CARASIL、年龄相关性黄斑变性7型、关节炎等,其中HTRA1基因纯合突变可导致CARASIL,特点是青少年起病,头部MRI可见脑深部白质病变,最常见的初始临床表现为痉挛性步态,在随后的5~20年可进展并逐渐出现其他神经系统症状。2015年,Verdura等首次报道HTRA1基因杂合突变所致的显性遗传性CSVD,且在欧洲不明原因家族性CSVD患者中,近5%不明原因家族性CSVD与HTRA1基因杂合突变有关。汉族家族性CSVD患者中,在排除NOTCH3基因突变的CSVD家系后,约5.61%携带HTRA1基因杂合突变。一项对181例疑似家族性CSVD患者进行的队列研究显示,HTRA1基因突变占5.5%(10/181)。有研究报道,HTRA1基因杂合突变成为CADASIL的第二常见原因,故其被称为CADASIL2型。与CARASIL不同,HTRA1基因杂合突变相关CSVD患者的临床特征为发病年龄较晚,40~60岁起病多见,男性患病率高,男性HTRA1基因杂合突变风险约为女性患者的2倍,且大部分患者表现为复发性卒中、认知障碍,部分可出现早发型脱发、脊椎病、偏头痛等,典型的影像学表现为FLAIR序列可见白质高信号,病灶常累及内囊及外囊。与CADASIL相比,HTRA1杂合突变相关CSVD患者脑白质病变累及前颞叶的比例较低。本例HTRA1杂合突变相关遗传性CSVD患者的外祖母、母亲、两位姨妈有脑梗死病史,均为女性,与既往研究报道的HTRA1基因杂合突变男性患者发病高于女性不一致。一项对4例HTRA1基因杂合突变相关CSVD患者的回顾性研究报道显示,首次卒中的中位年龄为51.25岁,中位随访时间6.5年,4例患者均发生复发性卒中,随访期间卒中发生的中位次数为5.5次,并在短时间内出现进行性肢体无力、双侧病理征、步态障碍、不同程度的认知障碍。本例患者首次卒中的年龄为51岁,本次出院后随访近10个月,未复发缺血性卒中,但仍需进行长期随访。
根据遗传变异分类标准与指南,本例患者的生物学致病等级为意义不明确,理由如下:(1)该突变最小等位基因频率<0.0005,属低频突变,在单碱基核苷酸多态性、千人基因组、千人南方、千人北方、基因组聚合数据库以及基因组聚合数据库的东亚数据库中未见收录,符合中等致病证据2;(2)经多种生物信息计算方法预测该突变对基因或基因产物有害或影响剪接的程度达到支持阈值水平,符合支持可能致病证据3。对本例患者的遗传模式进行分析,先证者为HTRA1c.947A>G杂合突变,符合常染色体显性遗传疾病的发病机制,但由于患者的父母已死亡,故无法判断遗传共分离情况。综合本例患者的临床主要表现、头部影像学特征等,其与HTRA1基因杂合突变导致的常染色体显性遗传性CSVD临床表现相符合。因此,依据生物学致病等级、遗传模式、临床匹配,推测HTRA1基因突变的危害性与患者表型存在相关性。
HTRA1基因杂合突变导致遗传性CSVD的发病机制尚未明确,目前推测可能与HTRA1基因杂合突变使蛋白酶的活性下降相关。本例患者蛋白结构预测的结果提示,HTRA1c.947A>G杂合突变导致第316位的酪氨酸突变为半胱氨酸,该突变位于编码丝氨酸蛋白酶的结构域(第204~364位氨基酸)。研究报道,位于该结构域其他位点的突变可导致遗传性CSVD,其原因为该结构域的突变可降低HTRA1蛋白酶的水解活性,由此推测HTRA1c.947A>G杂合突变也可能影响蛋白酶水解功能而致病。对HTRA1基因杂合突变导致CSVD患者进行尸检,显示其额叶脑白质存在局灶性髓鞘苍白,皮质下U型纤维未受累,颅内小血管内膜增生、内弹力板断裂、中膜平滑肌细胞广泛丢失、内膜及外膜纤维化,无淀粉样蛋白沉积,部分小血管显示畸形扩张或动脉瘤样改变,与既往报道的CARASIL病理特征相似,但严重程度较轻,因此二者可能存在相似的病理生理机制,但仍需大样本研究进一步验证。本例患者未进行HTRA1c.947A>G杂合突变后酶活性的体外试验,为本研究的局限性。
治疗方面,目前尚未见关于控制遗传性CSVD病情进展方法的报道,主要依靠经验行对症性治疗,对于应用抗血小板聚集药物的疗效,目前的研究结果并不一致。
综上,对于疑似遗传性CSVD的患者在进行NOTCH3基因筛查同时,需考虑存在HTRA1基因杂合突变的可能。临床实践中,神经科医师应提高对HTRA1基因杂合突变致遗传性CSVD的认识,合理借助基因检测方法,筛选CSVD高危家族患者并进一步指导治疗。
作者:中国脑血管病杂志
版权声明:
本网站所有注明“来源:梅斯医学”或“来源:MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明“来源:梅斯医学”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言









#脑小血管病# #HTRA丝氨酸肽酶1#
60