
黑色素瘤NGS大panel检测识别更多潜在治疗靶点,常见驱动基因阴性患者也可能有靶向治疗机会
刚刚 苏州绘真医学 苏州绘真医学
本研究强调了皮肤与 A/M 黑色素瘤之间,以及 A/M 内不同解剖部位之间的新的分子差异,可能会改变这些罕见黑色素瘤的临床检测和治疗范式。
皮肤黑色素瘤的治疗取得了进展,但肢端和黏膜 (A/M) 黑色素瘤患者的治疗选择仍然有限且预后不佳。我们使用 Foundation One panel 对 156 例黑色素瘤(101 例皮肤、28 例肢端和 27 例黏膜)进行了癌症相关基因检测,在特定的 A/M 黑色素瘤解剖部位识别了新的、潜在可靶向的基因变异 (GA)。使用新型 A/M 黑色素瘤临床前模型,我们证明与皮肤黑色素瘤相关的几种 GA 和相应的致癌通路在 A/M 黑色素瘤中同样可靶向。其他变异,包括 MYC 和 CRKL 扩增,是 A/M 黑色素瘤所特有的,分别对 BRD4 抑制剂 JQ1 或 Src/ABL 抑制剂达沙替尼间接靶向敏感。我们进一步发现了新的、可操作的 A/M 特异性变异,包括一例黏膜黑色素瘤中的 NF2 失活融合,对达沙替尼有体内反应。本研究强调了皮肤与 A/M 黑色素瘤之间,以及 A/M 内不同解剖部位之间的新的分子差异,可能会改变这些罕见黑色素瘤的临床检测和治疗范式。
研究背景
过去十年,晚期皮肤黑色素瘤的治疗领域取得了巨大进步,这在很大程度上得益于针对 BRAF 突变的靶向抑制剂和免疫检查点阻断 (ICB) 疗法的成功。然而,对于患有罕见非皮肤类型黑色素瘤的晚期黑色素瘤患者,包括肢端和黏膜 (A/M) 和眼部黑色素瘤,预后仍然通常不佳。在白种人人群中,A/M 黑色素瘤占恶性黑色素瘤的不到 6%,但在典型皮肤黑色素瘤并不常见的亚洲人群中,A/M 黑色素瘤占黑色素瘤病例的一半以上。肢端黑色素瘤发生在无毛的皮肤表面,例如脚底、脚趾、手掌(非甲下,非 SU)或指甲/拇指甲下(甲下,SU)。它们通常与阳光照射无关;然而,在某些情况下,这些肿瘤具有 DNA UV 特征。相比之下,黏膜黑色素瘤在黏膜组织中发展,例如鼻咽(头颈部,HN)、外阴阴道(VV)和肛门直肠(AR)。与肢端黑色素瘤一样,黏膜黑色素瘤与阳光照射无关,缺乏经典皮肤黑色素瘤中常见的 DNA UV 特征。
先前的NGS研究发现了 A/M 和皮肤黑色素瘤之间的许多基因组差异。特别是,A/M 黑色素瘤没有皮肤黑色素瘤中出现的高肿瘤突变负荷 (TMB)。此外,致癌 BRAF V600 突变在 A/M 黑色素瘤中并不常见。与低 TMB 形成对比,A/M 黑色素瘤的基因组不稳定性较高,导致结构性基因组变异 (GA),例如扩增、缺失和复杂重排。虽然 A/M 的总体 GA 情况已经确定,但许多 A/M 肿瘤仍被归类为“广泛阴性”,这意味着它们缺乏常见的黑色素瘤驱动变异,没有已知的致癌驱动因素。考虑到它们常发生基因组重排,我们和其他团队在这些黑色素瘤中识别了可靶向激酶融合作为致癌驱动因素也就不足为奇了。最近,在黏膜黑色素瘤中发现了其他潜在的致癌驱动因素,例如 SPRED1 和 ERBB2 突变;然而,对于很大一部分 A/M 黑色素瘤,致癌驱动变异仍然未知。
皮肤黑色素瘤对 ICB 的反应率较高,这被认为与高 TMB 有关。不幸的是,A/M 黑色素瘤对 ICB 反应率较低,通常需要二线靶向治疗。皮肤黑色素瘤的靶向治疗主要使用针对 MEK 和突变型 BRAF V600 的抑制剂联合治疗。由于 BRAF V600 突变在 A/M 黑色素瘤中不太常见,因此这种治疗方法通常不适用。一些靶向疗法,如 KIT 抑制剂,已在一小部分 A/M 黑色素瘤中显示出临床益处,但对于转移性 A/M 黑色素瘤患者,靶向治疗选择仍然非常有限。先前的基因组研究强调了 A/M 黑色素瘤的潜在治疗方法。但很少有人评估治疗疗效,因为罕见黑色素瘤的临床前模型很少,而且几乎没有临床试验专门针对罕见黑色素瘤。
在本研究中,我们对 55 例 A/M 和 101 例皮肤黑色素瘤进行了癌症相关基因检测,以识别新的致癌驱动因素和治疗靶点。此外,我们开发了新的 A/M 患者来源的异种移植 (PDX) 模型和细胞系来评估治疗效果。我们发现 A/M 黑色素瘤中的部分变异基因与皮肤黑色素瘤相同。A/M 细胞系对先前针对皮肤黑色素瘤的疗法表现出一定的敏感性;然而,许多变异是 A/M 黑色素瘤和这些亚型中不同解剖部位所特有的。这些变异可能是新的治疗靶点,例如黏膜黑色素瘤中的 NF2 突变和融合事件。本研究突出了 A/M 黑色素瘤的新分子特征,这些特征进一步将其与皮肤黑色素瘤区分开来,为开发专门针对罕见黑色素瘤患者的诊断panel和临床试验提供了新的靶点和先例。
研究结果
皮肤和A/M黑色素瘤的靶向基因检测
使用“Foundation One”panel对来自 156 例黑色素瘤成人患者的组织样本进行了靶向基因检测,其中包括 101 例皮肤黑色素瘤患者和 55 例肢端或黏膜 (A/M) 黑色素瘤患者(图 1A)。患者平均年龄为 61.7 岁,39% 为女性,63% 的组织来自原发性肿瘤(图 1A)。皮肤或肢端黑色素瘤的这些特征没有显著差异。黏膜黑色素瘤患者的平均年龄(68.0,p < 0.04)和女性患者比例(70%,p < 0.027)略高(图 1A)。
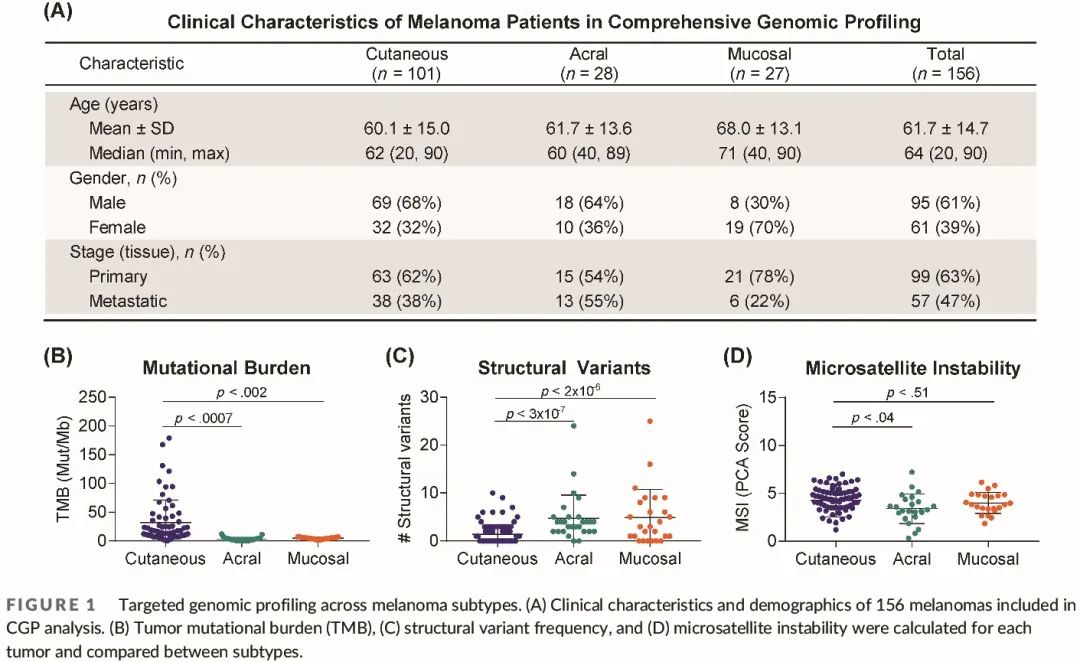
图1
对于每个样本,生成了 399 个癌症相关基因外显子和其中 28 个基因的选定内含子的杂交捕获下一代测序 (NGS) 数据。正常对照组织很少包括在临床分子检测中。因此,在数据分析过程中去除了常见的胚系 SNP,任何剩余的 SNP 都代表癌症相关基因中可能具有临床重要性的罕见变异。分析数据以确定肿瘤突变负荷 (TMB)、微卫星不稳定性 (MSI) 和四类 GA(碱基替换、插入/缺失、拷贝数变异和基因重排)。与之前的基因组研究结果一致,A/M 黑色素瘤的 TMB 显著低于皮肤黑色素瘤,而 A/M 黑色素瘤的结构变异(基因扩增、缺失、重排和融合)总数显著高于 A/M 黑色素瘤(图 1B、C)。皮肤黑色素瘤通常为“微卫星稳定”,MSI 频率较低,我们发现本研究中的所有黑色素瘤均微卫星稳定(图 1D)。
各黑色素瘤亚型和解剖部位的GA图谱
在 156 例黑色素瘤中发现了超过 3200 个 GA。皮肤黑色素瘤的 GA 平均频率(24.4 个 GA/患者)高于肢端(12.8 个 GA/患者,p < 0.002)或黏膜黑色素瘤(14.5 个 GA/患者,p < 0.007)。在分析的 399 个基因中,99 个(25%)在至少 5% 的 A/M 黑色素瘤中存在 GA(图 2)。在这 99 个基因中,超过一半(54%)也在 >5% 的皮肤黑色素瘤中存在 GA,其中包括已知的黑色素瘤相关基因,如 BRAF、NRAS、NF1、CDKN2A 和 PTEN(图 2)。46 个基因对 A/M 黑色素瘤更具特异性。这些基因包括 SF3B1、KIT、KRAS、MYC、HIST1H1C、PAG1 和 PCLO,它们在 >10% 的 A/M 中以及不到 4% 的皮肤黑色素瘤中存在 GA。这 46 个基因中有 18 个 (39%) 在皮肤黑色素瘤中没有 GA。这些基因包括参与 Notch 信号通路 (NOTCH4、MIB1)、表观遗传修饰 (JARID2、SETBP1)、DNA 修复 (CUX1、RAD21) 和抗原呈递 (B2M) 的基因。总之,A/M 黑色素瘤 GA 中有一部分与皮肤黑色素瘤重叠,但近一半的变异基因是 A/M 黑色素瘤特有的。
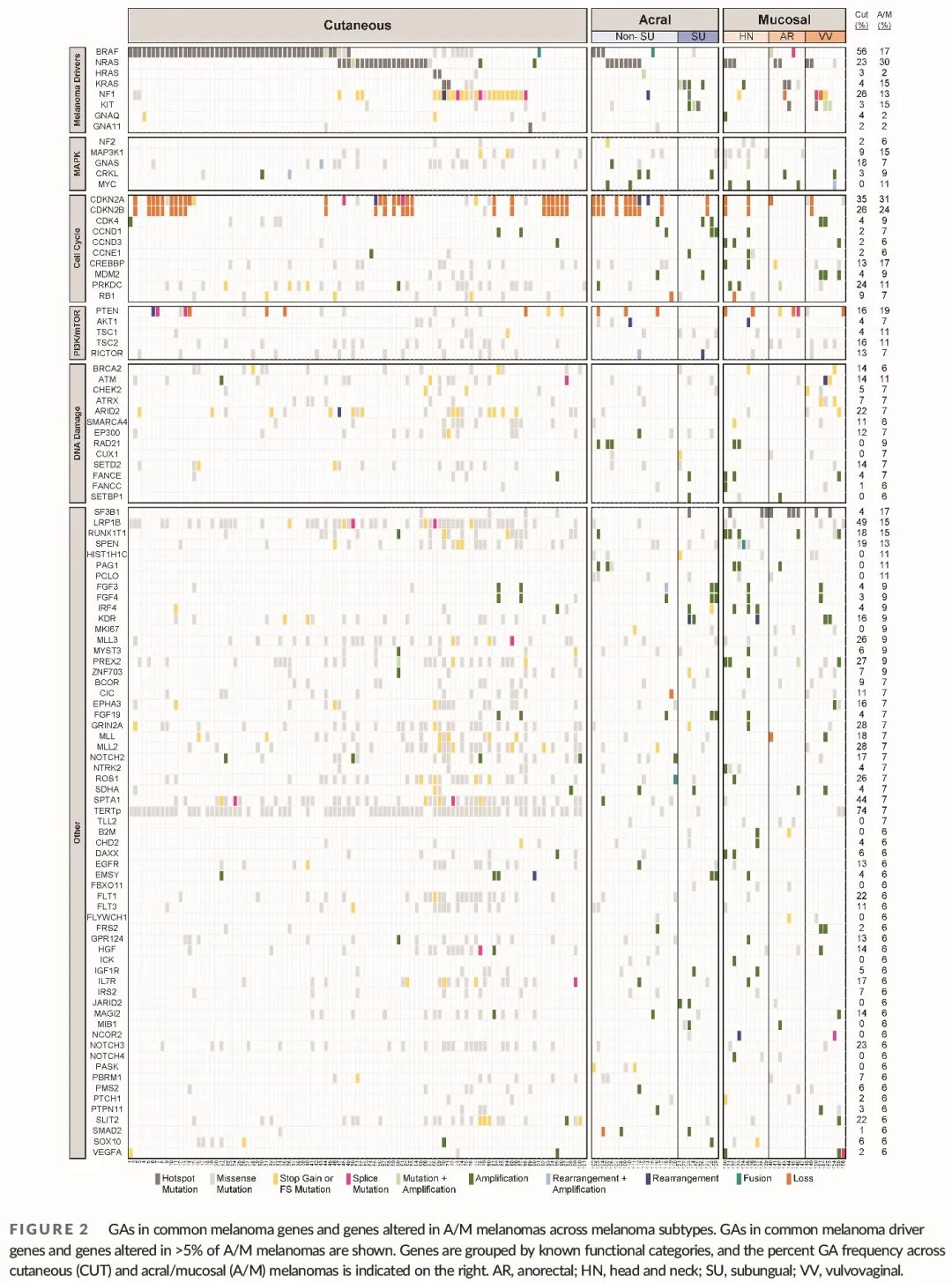
图2
各黑色素瘤亚型和解剖部位的常见黑色素瘤驱动基因分析
分析了 8 个常见黑色素瘤驱动基因 (BRAF、NRAS、HRAS、KRAS、NF1、KIT、GNAQ 和 GNA11) 的 GA 频率。正如预期的那样,皮肤黑色素瘤的 BRAF V600 突变频率较高 (图 3A)。BRAF V600 突变在肢端黑色素瘤中的发生率为 <10% (非 SU 中为 17%,SU 中为 0%),在黏膜黑色素瘤中未检测到。然而,7%–8% 的 A/M 黑色素瘤存在其他的 BRAF GA,包括非 V600 变异和 DGKI-BRAF 融合 (图 3A、B)。与皮肤黑色素瘤相比,A/M 黑色素瘤主要由 RAS 致癌基因 GA 驱动(皮肤黑色素瘤为 25%,A/M 为 38%–41%)(图 3A)。NRAS GA 在除 SU 黑色素瘤外的所有三种亚型中都很常见,SU 黑色素瘤往往携带 KRAS 变异。有趣的是,皮肤和肢端黑色素瘤常见 NRAS Q61K 和 Q61R 突变,而黏膜黑色素瘤携带 NRAS 和 KRAS Q61H 突变(图 3C)。
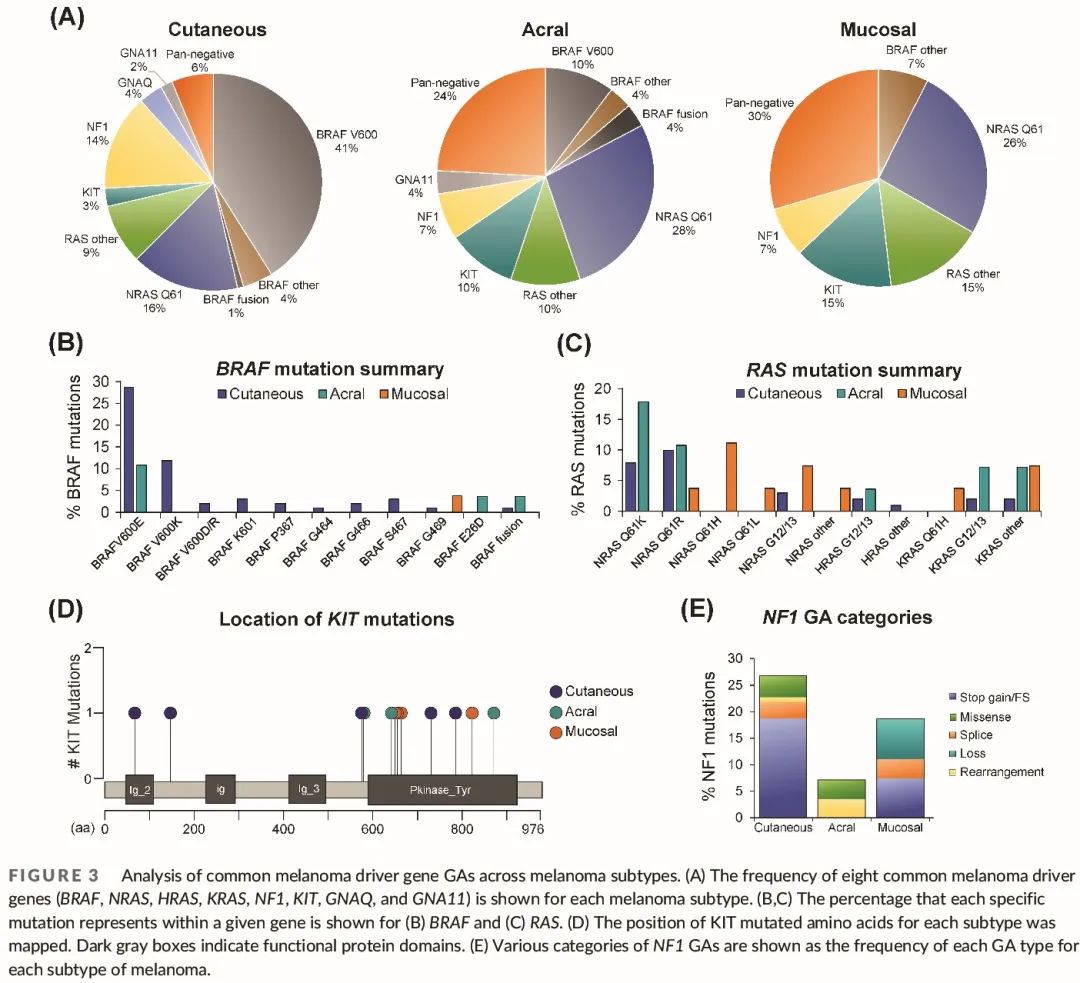
图3
KIT 是 A/M 黑色素瘤中突变频率第二高的黑色素瘤驱动基因(图 3A)。与既往研究一致,我们发现 KIT 突变主要发生于 SU 黑色素瘤和外阴阴道 (VV) 黏膜黑色素瘤。KIT 突变可位于整个基因不同位置,但大多聚集在激酶结构域内或附近(图 3D)。在3个皮肤黑色素瘤 KIT 突变中,只有1个 (Q575) 位于已知的致癌热点。相比之下,A/M 黑色素瘤中 5/7 的KIT 突变(L576P、V559A、K642E、N655K 和 N822K)是致癌的。我们证实 KIT 非热点突变是体细胞突变,不存在于患者胚系 DNA 中,但未来需要进行研究来确定这些突变是否是激活 KIT 的致癌驱动因素。
NF1 GA 在 A/M 黑色素瘤中的频率低于皮肤黑色素瘤(图 3A),但在 SU(44%)和肛门直肠 (AR) 黏膜(38%)黑色素瘤中的频率较高。NF1 是一种肿瘤抑制因子,在黑色素瘤和其他癌症中经常失活。大多数皮肤和 AR 黏膜 NF1 GA 是导致移码或终止密码子获得的突变(图 3E)。相比之下,SU 黑色素瘤经常发生结构变异,导致 NF1 缺失,例如重排和基因缺失(图 2 和 3E)。GNAQ 和 GNA11 GA 在超过 80% 的眼部黑色素瘤中被发现,但在皮肤和 A/M 黑色素瘤中不常见(图 3A)。
A/M黑色素瘤中潜在可靶向的重现性GA和通路
癌症中许多靶向治疗策略针对致癌驱动基因变异,但其他 GA 可能使其对特定靶向疗法敏感。为了识别 A/M 黑色素瘤中基于非驱动 GA 的其他潜在治疗策略,我们分析了这些黑色素瘤中的所有重现性突变和拷贝数 GA。在 >5% 的 A/M 黑色素瘤中发生 GA 的 99 个基因中,64 个基因在两个或更多样本中存在重现性 GA(图 2)。
最常出现的 GA 是 NRAS Q61 突变和 CDKN2A 缺失,在黏膜黑色素瘤(15%)中的发生率低于肢端黑色素瘤(32%)。随后是 PTEN 缺失和 SF3B1 R625 突变,绝大多数见于各黏膜黑色素瘤部位,除一例 SU 黑色素瘤。我们还观察到 15% 的黏膜黑色素瘤存在重现性 MAP3K1 S939C 突变,有趣的是,所有这些突变都发生在头颈部 (HN) 黑色素瘤中 (5/10,50%)。DNA 损伤修复通路基因有害突变 (获得终止密码子或移码突变) 并非重现性,但在 CM 中较常见 (图 2)。相比之下,这些突变在 A/M 黑色素瘤中很少见,除了 VV 黑色素瘤,其中 67% (6/9) 的肿瘤携带有害 GA。40 个基因发生重现性扩增 (阈值为 6 或更多拷贝),包括在至少 10% 的 A/M 中扩增的 22 个基因。这些扩增在皮肤黑色素瘤中不常见,发生率为 3% 或更低。最常见的扩增基因是 MYC,其次是 RUNX1T1、CDK4 和 CRKL。有趣的是,肛门直肠 (AR) 黏膜黑色素瘤的突变和扩增总体较少,包括较少的细胞周期基因扩增和 DNA 损伤修复基因变异(图 2)。
我们研究了在 64 个具有重现性变异的基因中富集的细胞区室、分子功能和生物过程。富集程度最高的7个生物过程涉及细胞周期/增殖或信号转导/RAS 信号传导过程。对 64 个具有重现性 GA 的基因进行 KEGG 功能通路富集分析,识别了 13 条在 A/M 黑色素瘤中显著富集的通路。这些通路包括 MAPK、细胞周期和 PI3K/AKT 通路,所有这些通路都可以作为治疗靶点。
在A/M黑色素瘤中靶向MAPK通路
为了基于靶向分析数据和基因本体分析评估治疗反应,我们从四个包含在分析中的 A/M 黑色素瘤肿瘤中衍生了细胞系。这些细胞系携带 MAPK、细胞周期、PI3K/AKT 和其他通路 GA(图 4A)。
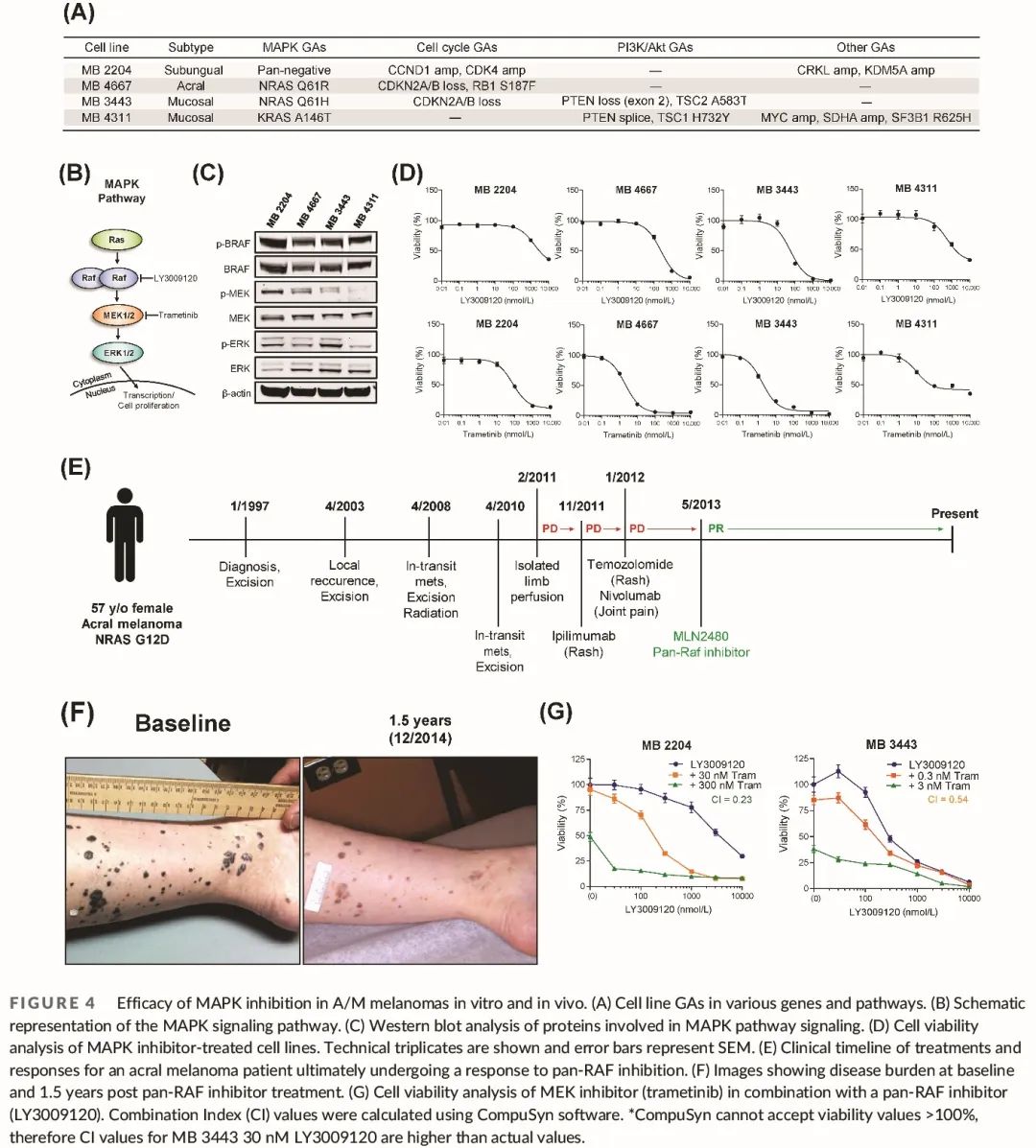
图4
A/M 黑色素瘤最常由 RAS 变异驱动,MAPK 通路是我们 GO 分析中富集的通路之一。四个细胞系中的两个(MB 3443 和 MB 4667)有 NRAS Q61 突变,一个细胞系(MB 4311)有 KRAS A146T 突变,一个细胞系(MB 2204)为广泛阴性(图 4A)。MAPK 通路激活状态通过蛋白质印迹法确定(图 4C),用 MEK 抑制剂(曲美替尼)或泛 RAF 抑制剂(LY3009120)治疗细胞(图 4D)。所有细胞系,包括广泛阴性 MB 2204 细胞系,对两种抑制剂均有显著反应,并且磷酸化 ERK 水平呈剂量特异性降低。MAPK 活性最高的细胞系(MB 3443)对两种药物的 IC50 值最低(图 4C)。
我们还观察到一例转移性肢端黑色素瘤患者对泛 RAF 抑制剂有反应(图 4E、F)。一例 57 岁的女性于 1997 年被诊断为足底肢端黑色素瘤。十年后,患者下肢多发移行转移。经过几次切除和放疗,疾病进展,患者接受了多种疗法治疗,包括隔离肢体灌注、化疗和免疫疗法,但均未成功。分子检测显示其黑色素瘤存在 NRAS G12D 突变。2013 年,患者进入泛 RAF 抑制剂 (MLN2480) 临床试验,剂量为每隔一天 200 mg,但出现了 3 级皮疹和 2 级血小板减少症。剂量减至每隔一天 140 mg,患者在治疗 6 周后获得部分缓解。由于持续出现不良反应,包括 1 级疲劳、肌痛、干眼、外周水肿和恶心,剂量随后降至每周 140 mg,副作用显著减少。患者仍在接受治疗,目前已超过 7.5 年,获得近乎完全缓解(图 4E、F)。
在皮肤黑色素瘤中,已证实 BRAF 和 MEK 抑制剂联合使用比单独使用其中一种药物更有效。此前,还观察到泛 RAF 和 MEK 抑制剂之间的协同作用。我们在一个 SU 黑色素瘤 (MB 2204) 和一个黏膜黑色素瘤 (MB 3443) 细胞系中测试了这种组合。发现当低剂量曲美替尼添加到泛 RAF 抑制剂中时没有协同作用,而高剂量的曲美替尼导致两种药物之间产生协同作用 (图 4G)。随着泛 RAF 抑制剂最近进入临床试验阶段,该联合疗法可能对 MAPK 通路变异的 A/M 患者有效。
在A/M黑色素瘤中靶向细胞周期通路
细胞周期通路是我们 GO 分析中最富集的通路 (图 5A)。我们在细胞系中在蛋白质水平验证了细胞周期 GA,确认了所有预期的蛋白表达变化,包括 MB 4667 和 MB 3443 中的 CDKN2A (p16) 缺失,以及 MB 2204 中的 CCND1 和 CDK4 扩增(图 5B)。我们用两种不同的 CDK4/6 抑制剂阿贝西利和哌柏西利治疗 MB 2204(CCND1 和 CDK4 扩增)SU 黑色素瘤细胞和 MB 3443(p16 缺失)黏膜黑色素瘤细胞(图 5C)。我们观察到两个细胞系之间的敏感性相似,尽管 CCND1 和 CDK4 扩增预测敏感性增强(图 5C)。最近的研究表明,CDK4/6 与 MEK 或泛 RAF 抑制剂联合使用对黑色素瘤具有协同作用。我们将阿贝西利与 MEK 抑制剂 (曲美替尼) 或泛 Raf 抑制剂 (LY3009120) 联合用于 MB 2204 细胞,但未观察到叠加或协同作用 (图 5D)。
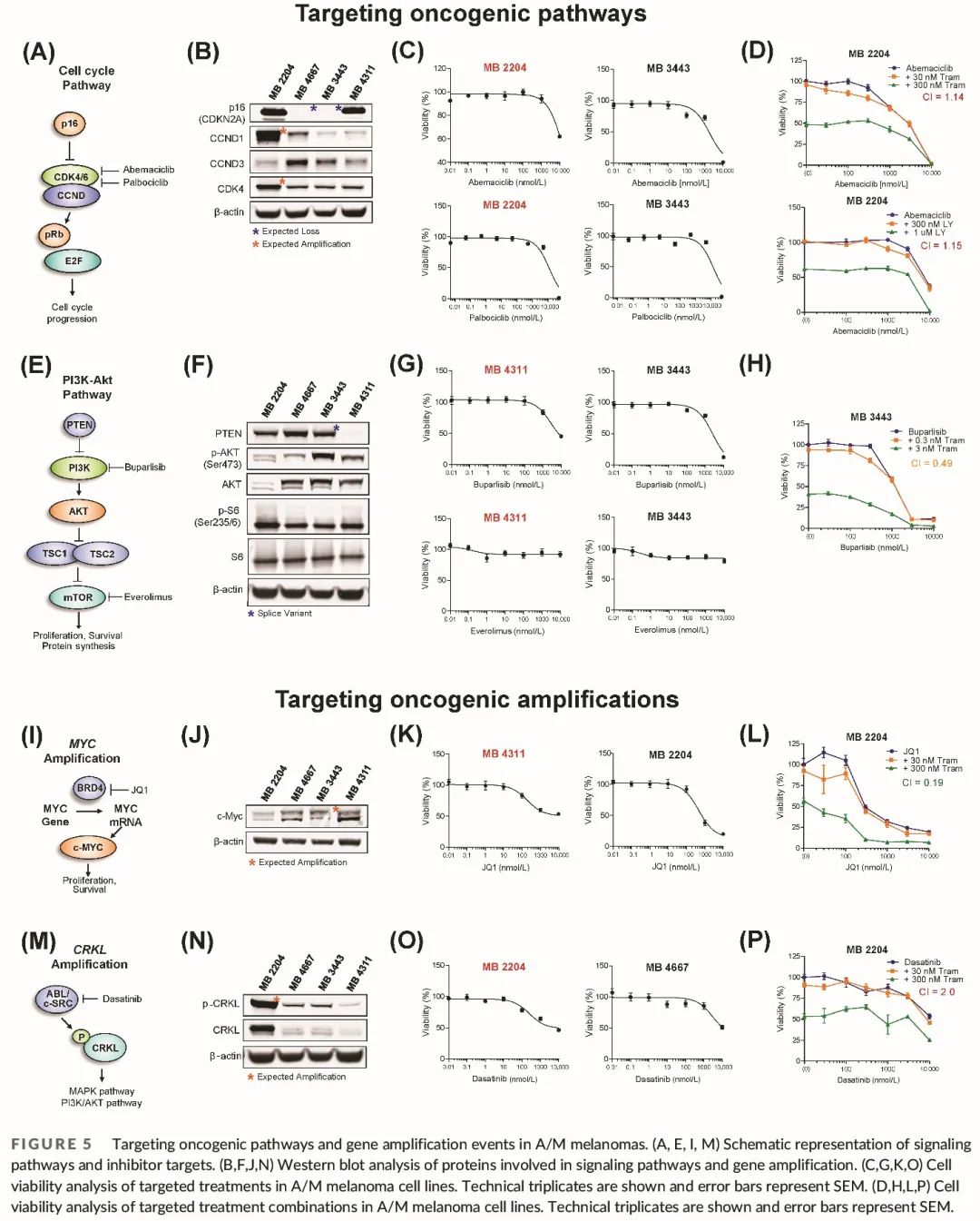
图5
在A/M黑色素瘤中靶向PI3K/AKT通路
PI3K/AKT 通路是我们 GO 分析中第二显著富集的通路 (图 5E)。我们验证了 TSC1 和 TSC2 (PI3K/AKT 通路的负调节因子) 突变,观察到MB 4311 细胞系中 PTEN 蛋白缺失,存在 PTEN 剪接变异 (图 5F)。在仅缺少外显子 2 而其余基因完整的 MB 3443 细胞系中未观察到 PTEN 蛋白完全缺失。我们用泛 PI3K 抑制剂buparlisib或下游 mTOR 蛋白抑制剂(依维莫司)治疗 MB 4311(PTEN 缺失)和 MB 3443(PTEN 剪接,TSC2 突变)黏膜黑色素瘤细胞(图 5G)。对buparlisib有反应,包括 AKT 磷酸化降低,在微摩尔范围内,与之前在黑色素瘤中的报告一致,但细胞对依维莫司没有反应(图 5G)。MEK 抑制剂和 PI3K 抑制剂联合治疗(之前报道在黑色素瘤中具有协同作用)在 MB 3443 细胞系中显示出轻微的叠加作用,但没有协同作用(图 5H)。
在A/M黑色素瘤中间接靶向基因扩增事件
MYC 是 A/M 黑色素瘤中最常见的扩增基因之一。已知 MYC 是一个致癌基因,其转录由表观遗传修饰因子 BRD4 促进(图 5I)。我们证实黑色素瘤细胞系 MB 4311 中 MYC 扩增导致蛋白质表达增加(图 5J)。为了间接靶向 MYC,我们用 JQ1(一种已知可降低 MYC 转录的 BRD4 抑制剂)治疗 MB 4311(MYC 扩增,MYC 高表达)黏膜黑色素瘤细胞和 MB 2204(无 MYC 变异,MYC 低表达)SU 黑色素瘤细胞(图 5I-K)。两个细胞系均对 JQ1 治疗有反应(图 5K),但 MB4311(MYC 扩增)细胞更敏感且 IC50 值较低(图 5K)。将 MEK 抑制剂(曲美替尼)加入 JQ1 可增强 MB 2204 肢端黑色素瘤细胞的反应,提示这种联合方法可能更有效地靶向 MYC 表达较低的细胞(图 5L)。
最后,我们分析了对靶向 CRKL 表达的反应。CRKL 是一种衔接分子,被 c-SRC磷酸化(间接通过 p130Cas),导致 MAPK 和 PI3K/AKT 通路激活(图 5M)。与无 CRKL 扩增的其他细胞系相比,MB 2204(CRKL 扩增)肢端黑色素瘤细胞中的 CRKL 和磷酸化 CRKL 蛋白表达显著较高(图 5N)。为了间接靶向 CRKL,我们用 c-SRC 抑制剂达沙替尼 (图 5M-O) 治疗 MB 2204 (CRKL 扩增,CRKL 高表达) 和 MB 4667 (CRKL 低表达) 肢端黑色素瘤细胞,达沙替尼降低了磷酸化 CRKL 水平,并且之前已被证实可用于治疗 CRKL 扩增的癌症。具有 CRKL 扩增的 MB 2204 细胞比 MB 4667 细胞更敏感 (IC50 值分别为 254 nM 和 3.1 μM)。由于达沙替尼还靶向其他激酶,包括各种 SRC 家族成员,我们用更特异性的 SRC 抑制剂tirbanibulin治疗细胞。与达沙替尼反应一致,我们发现具有 CRKL 扩增的 MB 2204 细胞比 MB 4667 细胞更敏感(IC50 值分别为 24 nM 和 >10 μM)。与 MEK 抑制剂(曲美替尼)联合使用未改善 MB 2204 对达沙替尼的反应(图 5P)。总之,这些数据表明,具有 MYC 或 CRKL 扩增和过表达的 A/M 黑色素瘤肿瘤可能分别对使用 JQ1 或达沙替尼的间接靶向敏感。
在广泛阴性黑色素瘤中识别新的潜在致癌驱动因素
大约四分之一 (24%–30%) 的 A/M 黑色素瘤是“广泛阴性”的,这意味着它们没有常见的黑色素瘤驱动基因 GA(图 3A)。因此,我们分析了广泛阴性黑色素瘤的基因组图谱,看是否可以识别潜在的其他致癌驱动变异。我们在 16/26 (62%) 的广泛阴性黑色素瘤中发现了 GA,它们可以作为潜在的驱动因素。其他驱动 GA 包括 MAPK 相关、受体酪氨酸激酶 (RTK) 和信号分子基因突变、融合和剪接变异(图 6A、B)。
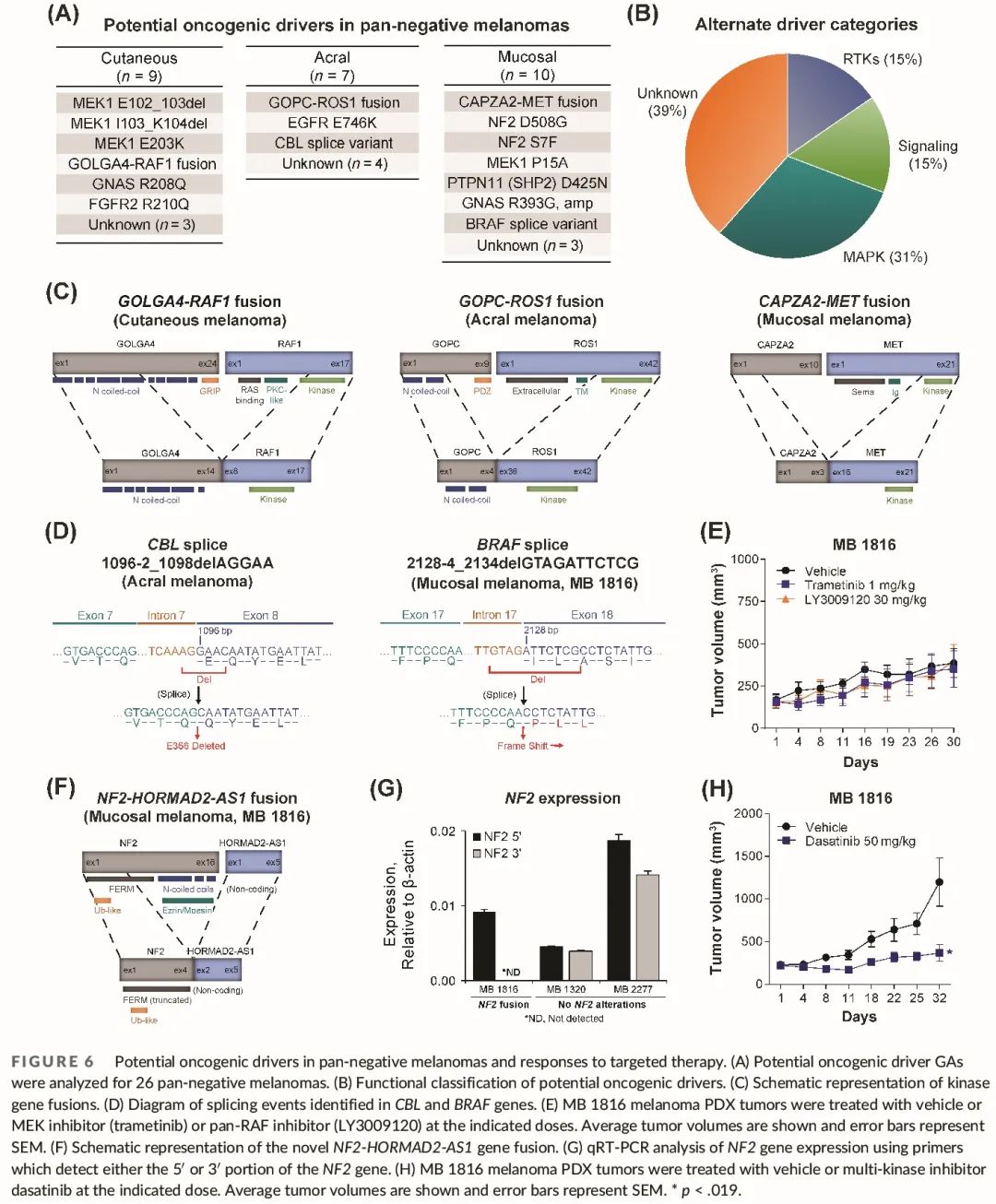
图6
MAP2K1 (MEK1) 突变最为常见,存在于三分之一 (3/9) 的皮肤黑色素瘤,包括2例碱基对缺失 (E102_I103del、I103_K104del) 和1例 E203K 突变。这些是已知的 MEK1 激活变异,可使肿瘤对 MEK 抑制剂疗法敏感。相比之下,MEK1 突变在 A/M 黑色素瘤中较少见,但我们在一例黏膜黑色素瘤中发现了功能意义未知的 MEK1 P15A 突变。识别了两种致癌 RTK 突变(FGFR2 R210Q、EGFR E746K),并且发现信号分子频繁突变,包括 PTPN11(SHP2)、GNAS(GNAQ 和 GNA11 的家族成员)和 NF2(NF1 的密切相关但不同的家族成员)(图 6A)。PTPN11 D425N 是一种已知的致癌突变,而 GNAS 突变尚未报道,但位于已知的热点致癌区域。在 20%(2/10)的广泛阴性黏膜黑色素瘤中发现了新的 NF2 突变,并且其中一例患者(携带NF2 S7F)有胚系 DNA 来确认该突变是体细胞突变(图 6A)。与 NF1 一样,NF2 抑制 RAS 活性。然而,NF2 还抑制 Hippo 信号通路,这会影响治疗 NF2 缺失驱动肿瘤的策略。
除了突变之外,还发现了三种致癌激酶基因融合,每种亚型各发现一种融合(图 6C)。在皮肤黑色素瘤中发现了 GOLGA4-RAF1 融合,既往研究显示,该融合对 MEK 抑制剂高度敏感。在肢端黑色素瘤患者中发现了 GOPC-ROS1 融合,我们之前报告过该患者对 ROS1 靶向治疗有非常好的反应。最后,在黏膜黑色素瘤中检测到并验证了 CAPZA2-MET 融合,据我们所知,这是该亚型中首次报道 MET 融合(图 6C)。先前已报道了具有 CAPZA2-MET 融合的癌症对 MET 抑制剂有反应。总体而言,超过 10% 的广泛阴性黑色素瘤具有可操作的激酶融合。
在2例广泛阴性 A/M 黑色素瘤中发现了潜在的致癌剪接事件。我们在肢端黑色素瘤中检测到 CBL 剪接事件,该事件导致 E366 氨基酸缺失(图 4D)。CBL 是一种肿瘤抑制泛素化酶,可促进许多致癌 RTK(包括 KIT)的泛素化和降解。我们还在一例黏膜黑色素瘤中发现了一种新的 BRAF 剪接事件(图 6D)。该事件导致最后一个 BRAF 外显子(外显子 18)开头的移码,从而导致无义和截短的 BRAF C 端。我们从该黏膜黑色素瘤开发了一个患者来源的异种移植 (PDX) 肿瘤(MB 1816),发现与没有该剪接事件的其他 A/M 黑色素瘤 PDX 肿瘤相比,其磷酸化 BRAF 水平升高。我们假设 MB 1816 肿瘤可能对 MAPK 抑制剂敏感;然而,在治疗肿瘤时没有观察到对 MEK 或泛 RAF 抑制剂的反应(图 6E)。
由于 MB 1816 黏膜黑色素瘤 PDX 模型不依赖于 MAPK 信号传导,我们对 MB 1816 PDX 模型进行了 RNA 测序,以寻找其他潜在的致癌驱动因素。令人惊讶的是,我们发现了一种以前未描述过的融合,涉及 NF2 抑癌基因和非编码反义转录本(图 6F)。该融合 NF2-HORMAD2-AS1 导致 NF2 C 端截断和 30 个基因表达缺失(图 6G)。有研究表明,NF2 缺失或失活驱动的肿瘤对达沙替尼敏感。我们用达沙替尼治疗 MB 1816 PDX 模型,观察到肿瘤生长显著减少(图 6H)。
讨 论
罕见的非UV相关黑色素瘤,包括肢端和黏膜 (A/M) 黑色素瘤,是侵袭性恶性肿瘤,诊断时通常处于晚期,预后一般比皮肤黑色素瘤差。虽然近年来皮肤黑色素瘤的治疗取得了重大进展,但这些新疗法对肢端和黏膜黑色素瘤的益处有限。在这项研究中,我们在来自不同解剖部位的 55 例 A/M 黑色素瘤中识别了癌症基因特异性 GA,并将这些 GA 与皮肤黑色素瘤进行了比较。我们关于 A/M 黑色素瘤的基本基因组特征和常见黑色素瘤驱动突变的数据与之前的基因组研究一致。此外,我们基于 A/M 黑色素瘤独特的分子特征和与皮肤黑色素瘤共同的通路,确定了新的致癌驱动变异和治疗策略。
本研究以及其他研究表明,RAS GA 是 A/M 黑色素瘤中最常见的致癌驱动因素。大多数特定的 RAS GA(NRAS Q61 突变)也存在于皮肤黑色素瘤,但频率较低。A/M 黑色素瘤中 RAS 基因经常发生扩增,而这在皮肤黑色素瘤中很少见。在我们的数据集中,NRAS Q61H 突变是黏膜亚型所特有的。对于不同的 NRAS Q61 和 G12/13 突变,观察到了不同程度的激活,需要进一步研究来确定 Q61H 突变在黏膜黑色素瘤中的潜在意义。
不幸的是,在各癌症中成功靶向 RAS 驱动的肿瘤是临床上的一大障碍。RAS 强烈激活 CRAF,CRAF 具有许多独立于其下游靶点MEK激酶激活的功能。因此,尽管体外数据令人鼓舞,但 RAS 驱动的肿瘤在临床上对单药 MEK 抑制剂的反应并不理想。近年来,已经开发出针对所有 RAF 蛋白(包括 CRAF)的“泛 RAF”抑制剂。在细胞系和小鼠模型中这些抑制剂可以成功治疗 RAS 和 BRAF V600 驱动的癌症。研究还证明了与 MEK 抑制剂联合使用的潜在协同作用。我们表明,单独使用泛 RAF 抑制剂,或尤其是与 MEK 抑制剂联合使用,可有效抑制 RAS 驱动的 A/M 细胞系的生长。我们还描述了一例参加泛 RAF 抑制剂临床试验的 NRAS G12D 阳性肢端黑色素瘤患者,转移性疾病持续近乎完全缓解超过 7.5 年。尽管泛 RAF 抑制剂似乎具有巨大的潜力,但在成功将这些药物广泛引入临床之前,还需要进一步研究。早期的单药临床试验令人失望,这些药物的生物利用度是目前的一个限制因素,但正在开发具有更好溶解度的泛 RAF 抑制剂。此外,并非所有具有 MAPK 变异的癌症都对泛 RAF 抑制有反应,例如我们的具有 BRAF 剪接变异的 MB 1816 黏膜黑色素瘤模型。然而,我们的数据显示这些抑制剂对罕见黑色素瘤有效,这使我们得出结论,开发更有效的下一代泛 RAF 抑制剂可能是罕见黑色素瘤治疗的重要下一步。
在 A/M 黑色素瘤中发现的许多 GA 与皮肤黑色素瘤不同,但通路分析显示了在皮肤黑色素瘤中也常常失调的多条致癌通路的富集。这些通路包括细胞周期和 PI3K/AKT 信号通路。近年来,细胞周期调节蛋白抑制剂(包括 CDK4/6)已进入临床,并在皮肤黑色素瘤和其他癌症(尤其是乳腺癌和卵巢癌)中显示出希望。我们证明 A/M 黑色素瘤对细胞周期抑制剂有体外反应,但未能看到与 MAPK 抑制剂联合使用的额外效果。尽管如此,先前研究表明 CDK4/6 抑制剂在 A/M 黑色素瘤的临床前模型中有反应,这进一步支持开发这些抑制剂用于治疗细胞周期异常的罕见黑色素瘤。A/M 黑色素瘤对 PI3K/AKT 抑制剂的体外反应有限,尽管与 MEK 抑制剂联合使用时有一些获益。
与皮肤黑色素瘤相比,A/M 黑色素瘤的突变较少,但基因组结构变异较多,包括扩增和复杂的基因重排。除了常发生细胞周期基因(例如 CCND1 和 CDK4)扩增外,我们和其他团队还发现了更特定于 A/M 黑色素瘤的扩增事件。本研究强调了 A/M 黑色素瘤中频繁的 MYC 和 CRKL 基因扩增,并表明在体外可以使用小分子抑制剂间接靶向具有这些变异的黑色素瘤。鉴于达沙替尼在临床上的可及性以及 JQ1 等 BRD4 抑制剂的快速发展,这些最终可能成为 A/M 患者的治疗选择。然而,达沙替尼和 BRD4 抑制剂的靶点很多,未来的研究需要验证 MYC 和 CKRL 对 A/M 黑色素瘤恶性程度和药物反应的影响。
最后,我们识别了几种新的潜在致癌驱动变异,这些变异是 A/M 黑色素瘤所特有的。这些驱动变异包括新的突变、剪接变异和基因融合。我们发现了未在皮肤黑色素瘤中见过的基因融合,反映了 A/M 黑色素瘤高度重排的特性。这些基因融合包括黏膜黑色素瘤中的 CAPZA2-MET 融合。我们还在黏膜黑色素瘤中发现了 NF2 基因的一种新型失活融合。该融合导致基因截断和 C 端 NF2 表达缺失。之前只有一项关于癌症中 NF2 融合的报告,是在脑膜瘤中发现的,是儿童期放疗的结果。本研究首次在未经治疗的肿瘤中识别了 NF2 失活融合作为潜在致癌驱动因素。黏膜黑色素瘤中的 NF2 失活融合以及 NF2 突变提示 NF2 变异可能是黏膜黑色素瘤特有的新型驱动基因型类别。与之前的数据一致,我们发现具有 NF2 失活融合的黏膜黑色素瘤 PDX 模型对达沙替尼(一种 FDA 批准的多酪氨酸激酶抑制剂)敏感。
本研究突出了不同 A/M 黑色素瘤解剖部位独特的基因组特征,这些特征不仅将它们与皮肤黑色素瘤区分开来,而且彼此之间也存在区别。由于 A/M 黑色素瘤很罕见,很少有研究全面比较 A/M 黑色素瘤内不同解剖位置之间的分子特征。在黏膜黑色素瘤中,外阴阴道 (VV) 和肛门直肠 (AR) 黑色素瘤通常被归为一类,称为“泌尿生殖系统”黑色素瘤。本研究识别了新的分子趋势,特别是在各黏膜黑色素瘤解剖部位。头颈部 (HN) 和 VV 黑色素瘤都经常出现细胞周期和 DNA 修复通路 GA,使它们倾向于某些靶向治疗,而这些 GA 在 AR 黑色素瘤中并不常见。进一步研究以扩大各黏膜黑色素瘤部位的样本数量对于验证这些临床相关发现至关重要。
尽管皮肤黑色素瘤和罕见黑色素瘤之间存在一些共同的变异和治疗方法,但这些共同点较少。本研究识别了 A/M 黑色素瘤新的基因组特征,并使用临床前模型探索治疗策略的有效性。A/M 黑色素瘤的临床前模型很少,而且在相关模型中测试的药物反应很少。本研究开发并利用了新的罕见黑色素瘤模型来证实它们确实对通过靶向基因组分析确定的药物敏感。我们还呈现了一例对泛 RAF 抑制剂有显著反应的肢端黑色素瘤患者的病例。根据这项研究,我们建议为 A/M 黑色素瘤建立特定的肿瘤检测panel和临床试验,以识别罕见黑色素瘤患者群体中这些可操作的治疗靶点并确定药物疗效。目前的范式是使用与皮肤黑色素瘤相同的药物治疗 A/M 黑色素瘤患者。总的来说,这已被证明是无效的。希望进一步了解这些独特肿瘤中存在的分子和 GA(如本研究发现的)能够带来新的有效治疗方法。
参考文献:
Turner JA, Van Gulick RJ, Robinson WA, Mughal T, Tobin RP, MacBeth ML, Holman B, Classon A, Bagby SM, Yacob BW, Hartman SJ, Silverman I, Vorwald VM, Gorden N, Gonzalez R, Gay LM, Ali SM, Benson A, Miller VA, Ross JS, Pitts TM, Rioth MJ, Lewis KD, Medina T, McCarter MD, Gonzalez R, Couts KL. Expanding the landscape of oncogenic drivers and treatment options in acral and mucosal melanomas by targeted genomic profiling. Int J Cancer. 2024 Jul 12. doi: 10.1002/ijc.35087. Epub ahead of print. PMID: 39001563.

作者:苏州绘真医学
版权声明:
本网站所有注明“来源:梅斯医学”或“来源:MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明“来源:梅斯医学”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言









#黑色素瘤# #NGS#
0