
Bluebird突破性基因疗法Lenti-D申请上市!改善致死罕见病CALD患者长期生存结局
2020-10-04 医药魔方 医药魔方
肾上腺脑白质营养不良(adrenoleukodystrophy,ALD)是一种致命性、神经退行性、X连锁隐性遗传病,发病率 0.5/10 万~1/10 万,多见于年轻男孩儿,影响全球大约21000例男
肾上腺脑白质营养不良(adrenoleukodystrophy,ALD)是一种致命性、神经退行性、X连锁隐性遗传病,发病率 0.5/10 万~1/10 万,多见于年轻男孩儿,影响全球大约21000例男性新生儿。
ALD发病原因是ABCD1 基因突变导致其编码的肾上腺脑白质营养不良蛋白(ALDP)结构改变或功能缺失,造成患者体内的极长链饱和脂肪酸(VLCFA) 不能进入过氧化物酶体进行β-氧化, 从而在血浆和组织细胞中蓄积,导致线粒体功能障碍,氧化应激加剧,生物膜功能破坏,最终引起神经系统脱髓鞘和肾上腺皮质功能减退等病理变化。
脑型肾上腺脑白质营养不良(cerebral adrenoleukodystrophy,CALD)是ALD最为严重的一种形式。大约40%的ALD男孩儿会进展为CALD,大多数患者负责思考和肌肉控制的神经功能严重丧失,如果不加以治疗,则进展迅速,最终死亡。CALD表现为6种主要人体功能障碍(MFDs),包括沟通障碍、皮质盲(双眼视觉完全丧失)、胃管喂食、完全失禁、轮椅依赖、自主运动完全丧失,严重损害患者的独立生活功能。
ALD目前尚无特效疗法,异体造血干细胞移植(allo-HSCT)是当前临床上ALD的标准治疗方式,可以纠正VLCFA代谢紊乱、改善临床症状,提高长期生存率。但allo-HSCT需要早期治疗才效果明显,而且受限于患者骨髓配型供体难以获得,另外allo-HSCT带来的免疫排斥反应、移植相关死亡、移植物抗宿主病风险都显着增高。基因疗法被认为是ALD最有潜力的治疗手段。
10月2日,Bluebird宣布欧洲药品管理局(EMA)已经正式受理其基因疗法elivaldogene autotemcel ( Lenti-D?,Eli-cel)用于治疗脑型肾上腺脑白质营养不良(CALD)的上市申请。今年7月,Lenti-D进入了EMA人用药品委员会(CHMP)的加速审评通道,可以让其欧盟上市申请的审批周期从210天缩短至150天。Bluebird计划2021年中向FDA提交Lenti-D的上市申请。
此前,Lenti-D治疗CALD曾获得FDA和EMA授予的孤儿药资格,并且于2018年5月获得了FDA授予的突破性疗法(BTD)资格,于2018年7月获得EMA授予的优先开发药物(PRIME)资格。Lenti-D还获得过FDA的罕见儿科疾病治疗药物资格认定,如果获批上市,Bluebird可以获得一张价值超过1亿美元的优先审评券。
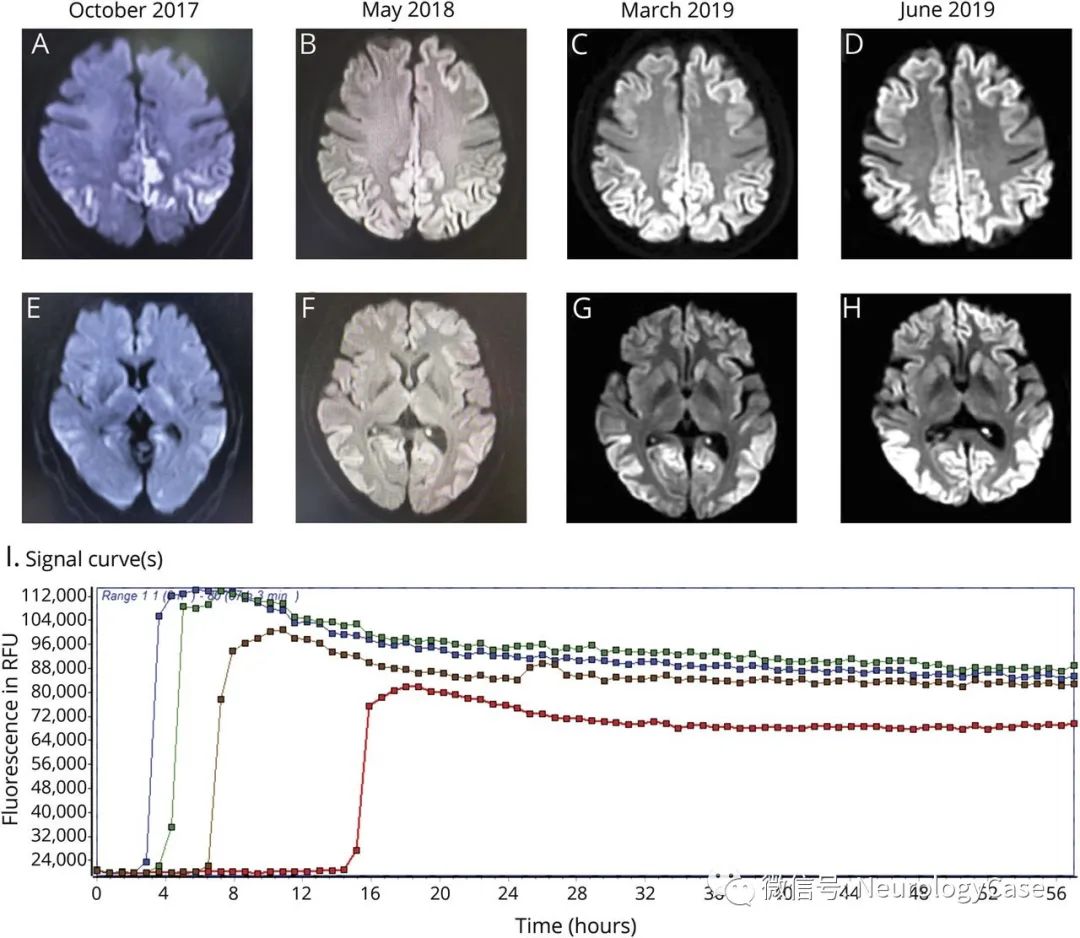
Lenti-D是从患者体内取出自体造血干细胞,在体外使用搭载人ABCD1基因的慢病毒转导修饰,然后回输给患者。功能基因在体内表达ALDP蛋白,就可以促进VLCFA降解,减轻其在大脑的积蓄。从FDA授予Lenti-D突破性疗法资格以及Bluebird此次向EMA提交上市申请所依据的II/III期ALD-102研究数据来看,Lenti-D对早期CLAD患者显示出了较好的延缓疾病进展的治疗潜力,且未见报道移植物抗宿主病、器官排斥或供体器官衰竭。
截至2020年1月,ALD-102研究共有32例患者接受Lenti-D治疗,中位随访时间为30个月(9.1~70.7个月),31例患者的神经功能评分稳定。需要特别指出的是,23例患者已经完成24个月随访,存活且无主要功能障碍(MRD-free)的比例达到87%(20/23)。这20例患者都继续进入到代号为LTF-304的长期随访研究中,其中14例已经完成至少4年的随访,10例已经完成至少5年随访。另外9例随访不足24个月的患者未表现出主要神经功能障碍。Bluebird还在开展一项代号为ALD-104的III期研究,评估CLAD患者先经过白消安和氟达拉滨清髓预处理后再接受Lenti-D治疗的疗效和安全性。
鉴于Lenti-D也是早期治疗效果更好,CALD的早期诊断很重要,因为在现有治疗条件下,患者的临床结局都取决于疾病发展的临床阶段。新生儿的ALD筛查是早期诊断以及成功治疗的关键因素。一旦新生儿被诊断患有ALD,定期进行MRI扫描以指示白质变化对于判断是否进展为CALD至关重要。
在美国,新生儿ALD筛查已经于2016年2月被列为推荐的通用筛查,目前在17个州和华盛顿特区均可进行,覆盖美国58%的新生儿。在美国境外,荷兰卫生已批准将ALD纳入其新生儿筛查计划。尽管大多数欧盟国家尚未实施ALD新生儿筛查,但是在这方面也在努力尝试推进。
作者:医药魔方
版权声明:
本网站所有注明“来源:梅斯医学”或“来源:MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明“来源:梅斯医学”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言






#申请上市#
59
#EBI#
65
#Bluebird#
60
#blue#
78
#罕见病##肾上腺脑白质营养不良#
185